

1 Introduction
Gene therapy is a promising approach for the treatment of cancer and inherited diseases [1]. In gene therapy, the most important is the vector which efficiently packages the DNA, carries it trough the membranes and deliver the gene to the nucleus for expression. An ideal vector should be highly efficient in delivering the gene in a target-specific manner, stable in vitro as well as in vivo, protect the gene from nuclease degradation, non toxic, non immunogenic and easily prepared in large quantities.
The most widely used types of vehicles for gene delivery are viral vectors and non-viral ones, such as liposomes.
Viral vectors are more efficient than their non-viral counterparts, but they have the disadvantage of being immunogenic, potentially mutagenic, with low viral titres and limited loading capacity in terms of DNA size.
Although less efficient in delivering gene fragments than virus, cationic liposomes have many important qualities, such as being much less or non-immunogenic and non-toxic. For this reason, we have decided to synthesise new cationic lipids for the preparation of liposomes able to vehicle nucleic acids for malign glioma gene therapy.
The most known cationic lipids, that formed cationic liposomes with a good delivering activity, are the N-[1-(2,3-dioleoyloxy)propyl]N,N,N-trimethyl ammonium chloride (DOTMA) [2], or 1,2 dioleoyloxy-3-(trimethylammonio) propane (DOTAP) [2] and 3β[N- (N′,N′-dimethylammino-ethane)carbamoyl]-cholesterol (DC-Chol) [3] (Fig. 1).
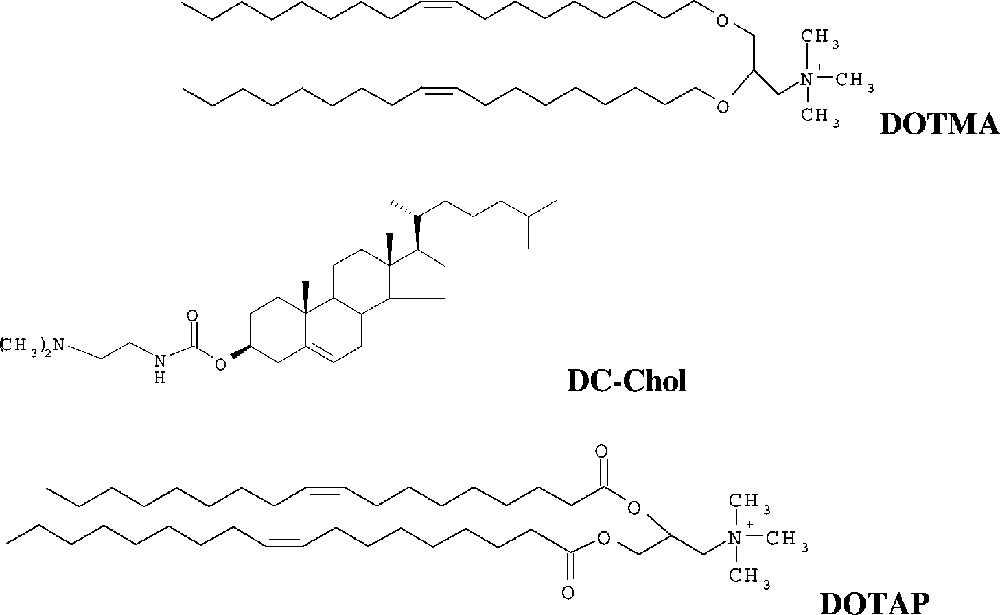
Structures of N-[1-(2,3-dioleoyloxy)propyl]N,N,N-trimethyl ammonium chloride (DOTMA) [2], 3β[N-(N′,N′-dimethylammino-ethane)carbamoyl]-cholesterol (DC-Chol.), and 1,2 dioleoyloxy-3-(trimethylammonio) propane (DOTAP).
DC-Chol has a tertiary amino group and a spacer that offers a good transfection activity, a stable carbamoyl bond that provides excellent stability and transfection efficiency as well as potential biodegradability, and hence reduced toxicity compared with the widely used cationic lipid with an ether bond. This means that is was deliberately designed with a relatively labile carbamoyl linker, which is not hydrolysed easily like the ester bond but, once inside the cell, is eventually biodegradable, probably by the cellular esterase.
2 Results and discussion
In an attempt to improve the delivering property of liposomes obtained from DOTAP mixed with dioleoyl-fosfatidyl-etanolamine (DOPE), we have synthesised analogues of DOTAP.
In all the structures that we designed, the bond between the lipophilic chain and the cationic head was preferably of carbammic nature. In fact, the relatively labile carbamoyl linker of Dc-Chol and analogues is one of the reasons for their good pharmaceutical characteristics [4].
The cationic lipids obtained are different, not only for the polar head groups, but also for the linkers.
We have used the modified Curtius reaction [5, 6] to obtain these lipids. The reaction consists in a one-pot reaction between the acyl chloride and the selected hydroxyl compound in the presence of sodium azide. In particular, we employed 3 bromo-1,2-propandiol 1 for the synthesis of a series of cationic glycerolipids. The contemporaneous presence of both primary and secondary alcoholic functions on the hydroxyl compound allows directing the reaction towards the mono- or di-carbamate, simply by varying the stoichiometry of the sodium azide.
In fact, the primary alcoholic function appears more reactive in this reaction and a mono-carbamate type 2 was obtained when equimolecular quantities of substrate and sodium azide are used. By a simple esterification reaction, the urethane/ester compound of type 3 is obtained. Finally a SN2 reaction gives the final product of type 4, as shown in Fig. 2.
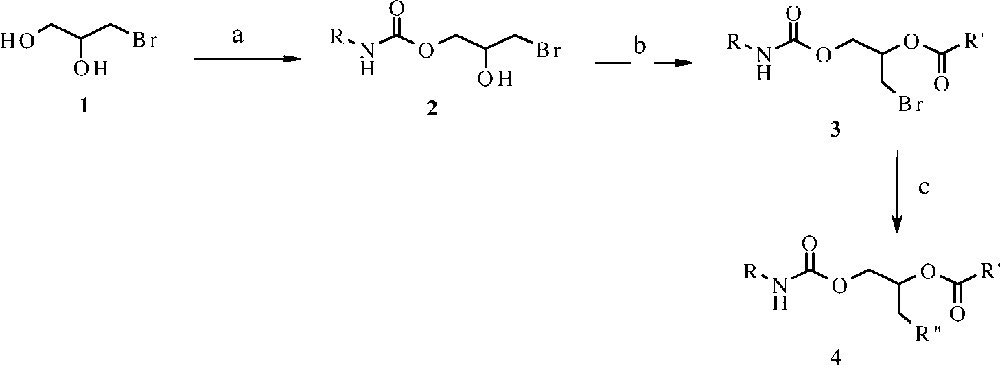
(a) 1.2 equiv NaN3, palmitoyl chloride or oleoyl chloride, benzyl-triethyl-ammonium chloride, toluene, 60 °C; (b) palmitoyl chloride or oleyl chloride, pyridine, DMAP, CH2Cl2; (c) morpholine or piperazine, CH3CN, 24 h. 2a: R = palmitoyl, yield 60%; 2b: R = oleyl, yield 52%; 3a: R = palmitoyl, R′ = palmitoyl, yield 90%; 3b: R = palmitoyl, R′ = oleyl, yield 85%; 3c: R = oleyl, R′ = oleyl, yield 87%; 3d: R = oleyl, R′ = palmitoyl, yield 85%; 4a: R = palmitoyl, R′ = palmitoyl, R′′= morpholine, yield 70%; 4b: R = palmitoyl, R′ = palmitoyl, R′′= piperazine, yield 65%; 4c: R = palmitoyl, R′ = oleyl, R′′= morpholine, yield 77%; 4d: R = palmitoyl, R′ = oleyl, R′′= piperazine, yield 69%; 4e: R = oleyl, R′ = oleyl, R′′= morpholine, yield 78%; 4f: R = oleyl, R′ = oleyl, R′′= piperazine, yield 67%; 4g: R = oleyl, R′ = palmitoyl, R′′= morpholine, yield 86%; 4h: R = oleyl, R′ = palmitoyl, R′′ = piperazine, yield 56%.
A fatty acid, the commercially available oleyl and palmitoyl chlorides are the hydrophobic moiety; as polar head, we have chosen an amine, in particular piperazine and morpholine. This synthetic methodology is shown to be versatile, because with simple synthetic steps and by modifying the reagents, it is possible to obtain a series of cationic lipids, with several polar head groups and linkers.
Both primary and secondary alcoholic functions are reactive when the quantities of sodium azide are doubled, affording in satisfactory yields the di-carbamate type 5. The cationic lipids types 6 are obtained using the previously described procedure, as reported in Fig. 3.

(a) 2.4 equiv NaN3, palmitoyl chloride or oleoyl chloride; (b) benzyl-triethyl-ammonium chloride, toluene, 60 °C, (c) morpholine or piperazine, CH3CN, 24 h, or dimethyl-amine, THF, 24 h. 5a: R = palmitoyl, yield 53%; 5b: R = oleyl, yield 46%; 6a: R = palmitoyl, R′ = morpholine, yield 60%; 6b: R = oleyl, R′ = morpholine, yield 57%; 6c: R = palmitoyl, R′ = piperazine, yield 52%; 6d: R = oleyl, R′ = piperazine, yield 72%; 6e: R = oleyl, R′ = dimethyl-amine, yield 82%; 6f: R = palmitoyl, R′ = dimethyl-amine, yield 76%.
Cationic lipids with a non-cyclic polar head group were also synthesised. We selected the dimethylamine instead of piperazine and morpholine and synthesized the corresponding lipids 6e and 6f. The synthetic procedure is the same for all cationic lipids synthesised and is reported in Fig. 3.
All before-reported cationic lipids form liposomes in combination with DOPE in 1:1 molar ratio, with the procedure reported in the ‘Experimental’ section.
The dimensions were measured by a light scattering apparatus and are suitable for transfection experiments [7]. In the dynamic light scattering technique, we measure the normalized time autocorrelation of the intensity of the scattered light which, for a dilute suspension of monodisperse particles decays exponentially with a decay rate G = 2 q2D, where q is the magnitude of the scattering wave vector and D the diffusion coefficient. The hydrodynamic radius of the diffusing particles is obtained through the Stokes–Einstein relationship. For a polydisperse system, when the single exponential decay is no longer valid, the size and size distribution of the diffusing particles were found using the standard data analysis program CONTIN, in terms of a continuous distribution of exponential decay times. A typical example of the autocorrelation function measured by dynamic light scattering and the CONTIN analysis giving the size distribution is shown in Fig. 4.
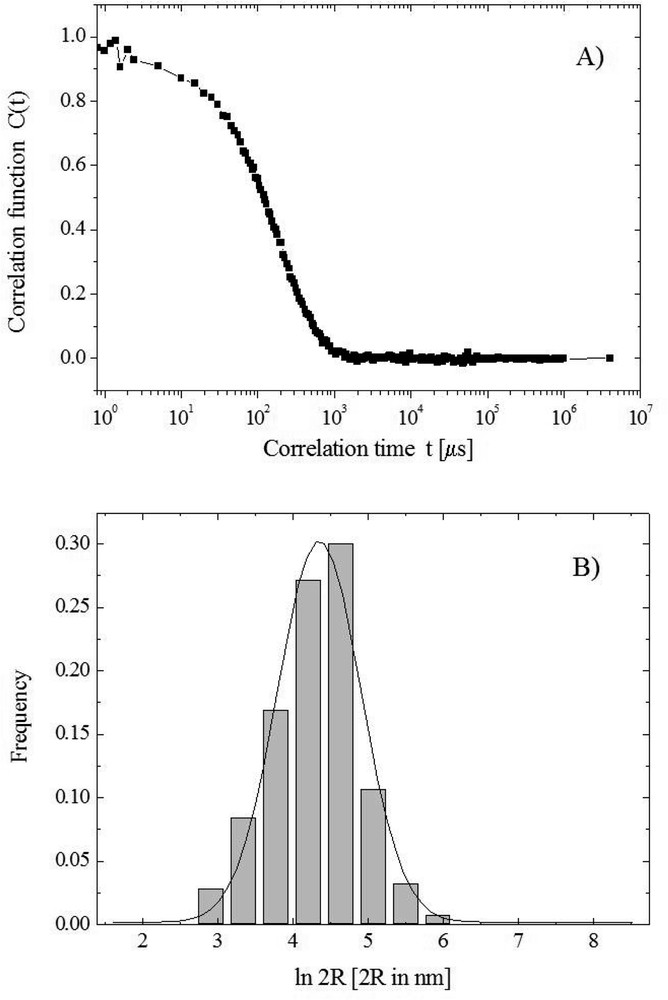
Autocorrelation function measured by dynamic light scattering and the CONTIN analysis.
For all the samples investigated, the relaxation time distribution gives a single population (monomodal distribution), with some degree of polydispersity (a measurement of the reduced second moment of the distribution), ranging from 0.08 to 0.13, as reported in Table 1.
Selected liposome characteristics.
| Lipid | Polar head group | Liposome mean diameter (nm) | Liposome polydispersity |
| 6c | Piperazine | 122 | 0.10 |
| 4d | Piperazine | 145 | 0.10 |
| 4c | Morpholine | 214 | 0.08 |
| 6b | Morpholine | 420 | 0.13 |
| 4e | Morpholine | 240 | 0.09 |
| 6e | Dimethyl-amine | 290 | 0.10 |
The cationic lipid 6e (code name: MM54) resulted to be the most interesting one for its transfection good properties [7] in C6 glyoma cells.
3 Experimental
3.1 General remarks
All reactions were carried out under dry argon using anhydrous solvents of Sigma-Aldrich. Glassware was flame dried prior to use. Commercial reagents were purchased from Sigma-Aldrich or Fluka and were used without further purification. Reactions were monitored by thin layer chromatography (TLC) on Merck silica gel plates (0.25 mm); spots were visualized using UV light or by spraying with phosphomolybdic reagent. Reaction temperature was measured externally. Flash chromatography was performed on Merck silica gel 60 (particle size 0.040–0.063 mm). Yields refer to chromatographically and spectroscopically (1H NMR) pure materials. NMR spectra were recorded in a CDCl3 solution on a Varian Gemini 200 spectrometer at room temperature. Chemical shifts are reported relative to the residual solvent peak (CHCl3: δH = 7.26, CDCl3: δc = 77.0).
3.2 Liposome preparation
Liposomes were prepared by the lipid hydration method. Appropriate amounts of lipids, in 1:1 molar ratio with DOPE, were dissolved in ethanol, followed by injection in sterile deionised water under vigorous stirring [8] to reach a final concentration of 1 mg ml–1, at a temperature above the phase-transition temperature. The resulting aqueous lipid mixture was sonicated at pulsed power mode followed by extrusion through polycarbonate filters of varying size. A homogeneous liposomal suspension of unilamellar vesicles was obtained, as reported in Fig. 1 and Table 1.
3.3 Dynamic light-scattering measurements
Light-scattering experiments were performed using a classic set-up with a correlator (BI9000AT, Brookhaven) operating with a logarithmic sample time spacing. The light source is a 10-mW He–Ne laser (wavelength 632.8 nm) and the measurements have been made at a scattering angle of 90°.
4 Synthesis of intermediate 2
The acyl chloride (1 mmol) and dry benzyltriethylammonium chloride (0.1 mmol) were added to dry toluene (2 ml) stirring for 5 min at 60 °C. Dry sodium azide (1.2 mmol) was added in portions (4 × 0.3 mequiv) over 1 h at 60 °C under stirring. The solution was kept for 15 min at 80 °C. The alcohol (5 mmol) was then added at room temperature and allowed to stand overnight. The solution was diluted with ethyl ether (50 ml), washed with water and dried with anhydrous sodium sulphate. Solvent removal gave the product that was purified on a column of flash silica gel, eluting with 0–2% MeOH in chloroform.
4.1 Product 2a
1H NMR δ: 3.15 (2H, bq, J = 6 Hz, CH2N), 3.40 (2H, m, CH2Br), 4.02 (1H, m, CHO), 4.16 (1H, m, CHOH), 5.60 (1H, m, NH). 13C–NMR CDCl3 200 MHz δ: 14.04, 18.08, 22.66, 26.80, 29.12, 29.47 (× 2), 29.70 (× 2), 29.85 (× 2), 31.92, 34.16, 41.23, 57.94, 66.42, 69.53, 156.90.
4.2 Product 2b
1H NMR δ: 3.19–3.23 (2H, bq, J = 12 Hz, CH2NH), 3.45–3.49 (2H, dd, J = 2.2 Hz, J′ = 6 Hz, CH2Br), 4.02–4.044 (1H, m, CHOH), 4.24–4.26 (2H, bd, J = 4 Hz, CH2OC=O), 4.88 (1H, m, NH), 5.33–5.38 (2H, m, CH = CH).
4.3 Synthesis of intermediate 3
To a solution of 2 (1 mmol) in CH2Cl2 anhydrous (1 ml) is added the acyl chloride (3 mmol), pyridine (3 mmol) and DMAP (0.1 mmol). The reaction was allowed to stand at room temperature overnight. Then solvents were evaporated in vacuo and the residue purified by flash chromatography (104–20% EtOAc in petroleum ether), to give 3.
4.4 Product 3a
1H NMR δ: 2.40 (1H, t, J = 8 Hz, CH2CO), 3.15 (2H, bq, J = 6 Hz, CH2N), 3.40 (2H, m, CH2Br), 4.20 (1H, m, CHOCONH), 4.10 (1H, m, CHOH), 4.80 (1H, m, NH), 5.20 (1H, m, CHOCO).
4.5 Product 3b
1H NMR δ: 2.30–2.39 (2H, m, CH2C=O), 3.142–3.17 (2H, m, CH2NH), 3.46–3.52 (2H, m, CH2Br), 4.24–4.28 (CH2OC=ONH), 4.80 (1H, m, NH), 5.20–5.22 (1H, m, CHOC=O), 5.31–5.36 (2H, m, CH = CH)
4.6 Product 3c
1H NMR δ: 2.37–2.40 (2H, m, CH2C=O) 3.19–3.23 (2H, bq, J = 12 Hz, CH2NH), 3.45–3.49 (2H, dd, J = 2.2 Hz, J’ = 6 Hz, CH2Br), 4.24–4.26 (2H, bd, J = 4 Hz, CH2OC=O), 4.88 (1H, m, NH), 5.09 (1H, m, CHOC=O), 5.33–5.38 (2H, m, CH = CH).
4.7 Product 3d
1H NMR δ: 2.30–2.39 (2H, m, CH2C=O), 3.12–3.18 (2H, m, CH2NH), 3.47–3.53 (2H, m, CH2Br), 4.25–4.30 (CH2OC=ONH), 4.80 (1H, m, NH), 5.20–5.22 (1H, m, CHOC=O), 5.32–5.37 (2H, m, CH = CH)
4.8 Synthesis of intermediate 5
The acyl chloride (1 mmol) and dry benzyltriethylammonium chloride (0.1 mmol) were added to dry toluene (2 ml) under stirring for 5 min at 60 °C. Dry sodium azide (2.4 mmol) was added in portions (4 × 0.6 mmol) over 1 h at 60 °C under stirring. The solution was kept for 15 min at 80 °C. The alcohol (5 mmol) was then added at room temperature and allowed to stand overnight. The solution was diluted with ethyl ether (50 ml) washed with water and dried with anhydrous sodium sulphate. Solvent removal gave the product, which was purified on a column of flash silica gel, eluting with 0–2% MeOH in chloroform.
4.9 Product 5a
1H NMR δ: 3.15 (4H, bq, J = 6 Hz, CH2N), 3.40 (2H, m, CH2Br), 4.02 (2H, m, CH2OCONH), 4.10 (1H, m, CHOH), 5.20 (1H, m, CHOCONH), 5.60 (1H, m, NH).
C13 CDCl3 200 MHz δ: 14.13 (× 2), 22.74 (× 2), 26.80 (× 2), 29.35 (× 2), 29.41 (x4), 29.64 (x4), 29.73 (x4), 30.08 (× 2), 30.85 (× 2), 31.98 (× 2), 41.23, 51.96, 63.95, 70.88, 76.50, 155.26, 166.90.
4.10 Product 5b
1H NMR δ: 3.19–3.23 (4H, bq, J = 12 Hz, CH2NH), 3.45–3.49 (2H, dd, J = 2.2 Hz, J′ = 6 Hz, CH2Br), 4.24–4.26 (2H, bd, J = 4 Hz, CH2OC=O), 4.88 (1H, m, NH), 5.20 (1H, m, CHOC=O), 5.33–5.38 (2H, m, CH = CH).
4.11 Synthesis of type-4 and type-6 lipids
To a solution of 3 (1 mmol) in CH3CN (10 ml), morpholine or piperazine (20 mmol) is added. The reaction mixture was refluxed overnight, concentrated in vacuo to remove CH3CN, and the residue was flash-chromatographed (chloroform for morpholine derivatives and 2% MeOH in chloroform for piperazine derivatives), to give 5.
To a solution of 5 (1 mmol) in THF (10 ml), 2 M dimethylamine in THF (20 mmol) is added. The reaction mixture was refluxed overnight concentrated in vacuo to remove THF, and the residue was flash chromatographed on 5% MeOH in chloroform.
4.12 Product 4a
1H NMR δ: 2.40 (1H, t, J = 8 Hz, CH2CO), 2.65 (4H, m, CH2N), 3.15 (2H, bq, J = 6 Hz, CH2N), 3.72 (4H, m, CH2O), 4.20 (1H, m, CHOCONH), 4.10 (1H, m, CHOH), 4.80 (1H, m, NH), 5.20 (1H, m, CHOCO).
4.13 Product 4b
1H NMR δ: 2.29–2.32 (2H, t, J = 6 Hz, CH2C=O), 2.60 (4H, m, CH2N), 3.19–3.22 (2H, bq, CH2NHC=O), 3.77 (4H, m, CH2O), 4.19–4.22 (2H, bq, CH2OC=O), 4.80 (1H, m, NH), 5.20 (1H, m, CHOC=O).
4.14 Product 4c
1H NMR δ: 2.29–2.33 (2H, t, J = 6 Hz, CH2C=O), 2.50–2.54 (4H, m, CH2N), 3.15–3.18 (2H, bq, CH2NHC=O), 3.67–3.72 (4H, t, J = 4 Hz, CH2O), 4.19–4.22 (2H, bq, CH2OC=O), 4.80 (1H, m, NH), 5.20 (1H, m, CHOC=O), 5.30 (2H, m, CH = CH).
4.15 Product 4d
1H NMR δ: 2.30–2.39 (2H, m, CH2C=O), 3.10 (2H, m, CH2N), 3.14–3.17 (2H, m, CH2NH), 3.40 (4H, m, CH2N), 3.62 (4H, m, CH2NH), 4.24–4.28 (CH2OC=ONH), 4.80 (1H, m, NH), 5.20–5.22 (1H, m, CHOC=O), 5.31–5.36 (2H, m, CH = CH).
4.16 Product 4e
1H NMR δ: 2.29–2.33 (2H, t, J = 6 Hz, CH2C=O), 2.50–2.54 (4H, m, CH2N), 3.15–3.18 (2H, bq, CH2NHC=O), 3.67–3.72 (4H, t, J = 4 Hz, CH2O), 4.19–4.22 (2H, bq, CH2OC=O), 4.80 (1H, m, NH), 5.20 (1H, m, CHOC=O), 5.30 (4H, m, CH = CH).
4.17 Product 4f
1H NMR δ: 2.30–2.39 (2H, m, CH2C=O), 3.10 (2H, m, CH2N), 3.14–3.17 (2H, m, CH2NH), 3.40 (4H, m, CH2N), 3.62 (4H, m, CH2NH), 4.24–4.28 (CH2OC=ONH), 4.80 (1H, m, NH), 5.20–5.22 (1H, m, CHOC=O), 5.31–5.36 (4H, m, CH = CH)
4.18 Product 4g
1H NMR δ: 2.29–2.33 (2H, t, J = 6 Hz, CH2C=O), 2.50–2.54 (4H, m, CH2N), 3.15–3.18 (2H, bq, CH2NHC=O), 3.67–3.72 (4H, t, J = 4 Hz, CH2O), 4.19–4.22 (2H, bq, CH2OC=O), 4.80 (1H, m, NH), 5.20 (1H, m, CHOC=O), 5.30 (4H, m, CH = CH).
4.19 Product 4h
1H NMR δ: 2.30–2.39 (2H, m, CH2C=O), 3.10 (2H, m, CH2N), 3.14–3.17 (2H, m, CH2NH), 3.40 (4H, m, CH2N), 3.62 (4H, m, CH2NH), 4.24–4.28 (CH2OC=ONH), 4.80 (1H, m, NH), 5.20–5.22 (1H, m, CHOC=O), 5.31–5.36 (2H, m, CH = CH).
4.20 Product 6a
1H NMR δ: 3.15 (4H, bq, J = 6 Hz, CH2NHCO), 2.60 (4H, m, CH2NH), 3.40 (4H, m, CH2N), 3.70 (4H, m, CH2O), 4.02 (2H, m, CH2OCONH), 4.10 (1H, m, CHO), 4.90 (1H, m, NH), 5.20 (2H, m, CHOCONH).
4.21 Product 6b
1H NMR δ: 3.15 (4H, bq, J = 6 Hz, CH2NHCO), 2.90 (4H, m, CH2NH), 3.20 (2H, m, CH2N), 3.40 (4H, m, CH2N), 4.02 (2H, m, CH2OCONH), 4.10 (1H, m, CHO), 4.90 (1H, m, NH), 5.20 (2H, m, CHOCONH).
4.22 Product 6c
1H NMR δ: 3.15 (4H, bq, J = 6 Hz, CH2NHCO), 2.60 (4H, m, CH2NH), 3.40 (4H, m, CH2N), 3.70 (4H, m, CH2O), 4.02 (2H, m, CH2OCONH), 4.10 (1H, m, CHO), 4.90 (1H, m, NH), 5.20 (2H, m, CHOCONH).
4.23 Product 6d
1H NMR δ: 3.15 (4H, bq, J = 6 Hz, CH2NHCO), 2.90 (4H, m, CH2NH), 3.20 (2H, m, CH2N), 3.40 (4H, m, CH2N), 4.02 (2H, m, CH2OCONH), 4.10 (1H, m, CHO), 4.90 (1H, m, NH), 5.20 (2H, m, CHOCONH).
4.24 Product 6e
1H NMR δ: 2.32–2.37 (2H, m, CH2N), 2.38 (6H, s, N (CH3)2), 3.13–3.16 (2H, bq, CH2N), 3.85–3.99 (2H, m, CH2OC=O), 5.20 (1H,m, CHOC=O), 5.30–5.36 (4H, m, CH = CH).
4.25 Product 6f
1H NMR δ: 2.36–2.39 (2H, m, CH2N), 2.41 (6H, s, N (CH3)2), 3.15–3.19 (2H, bq, CH2N), 3.92–3.99 (2H, m, CH2OC=O), 5.25 (1H,m, CHOC=O).
Acknowledgements
MIUR and CNR are acknowledged for financial support.