

One of the major challenges in synthetic organic chemistry is the design and execution of concise approaches and strategies to complex molecules by using reactions that rapidly lead to their carbon skeleton. In this context, we have explored the construction of molecules having diol and polypropionate units by tuning up regio-, chemo-, stereo- and enantioselective reactions.
Asymmetric C–C bond formation is an attractive synthetic strategy, as the connectivity of a carbon framework and its configuration are established simultaneously. The allylmetallation of aldehydes and ketones, leading to products with a maximum of two new stereocentres and versatile functionalities for further transformations is an important method for acyclic stereocontrol [1–5]. Efficient chirality transfer from allyl moieties has been obtained with chiral allylmetal reagents [6–13]. In the 1980s, organotitanium reagents opened new perspectives for high selectivity, as their structural variability allows the necessary adjustments for reactivity [14–19]. Duthaler and Hafner prepared a variety of such chiral organotitanium reagents and among them, chiral cyclopentadienyldialkoxytitanium (IV) complexes [20, 21]. They screened them for enantioselective allyltitanations and crotyltitanations and they discovered that the presence of the (R,R)-tartrate ligand in complex (R,R)-I and (R,R)-II allows the transfer of allyl and crotyl groups to aldehydes with high si face selectivity and that the presence of the (S,S)-tartrate ligand in complex (S,S)-I and (S,S)-II allows the transfer of allyl and crotyl groups to aldehydes with high re face selectivity [22, 23]. A wide range of homoallylic alcohols with high enantiomeric excess was obtained by using these complexes (Fig. 1).

Enantioselective allyl- and crotyltitanium complexes.
Nevertheless, it was desired to develop methodologies for the stereocontrolled obtention of 1,2- or 1,3-diol units as well as polypropionate units to prepare biologically active compounds such as LY 33 35 31, passifloricin A and discodermolide (Fig. 2).
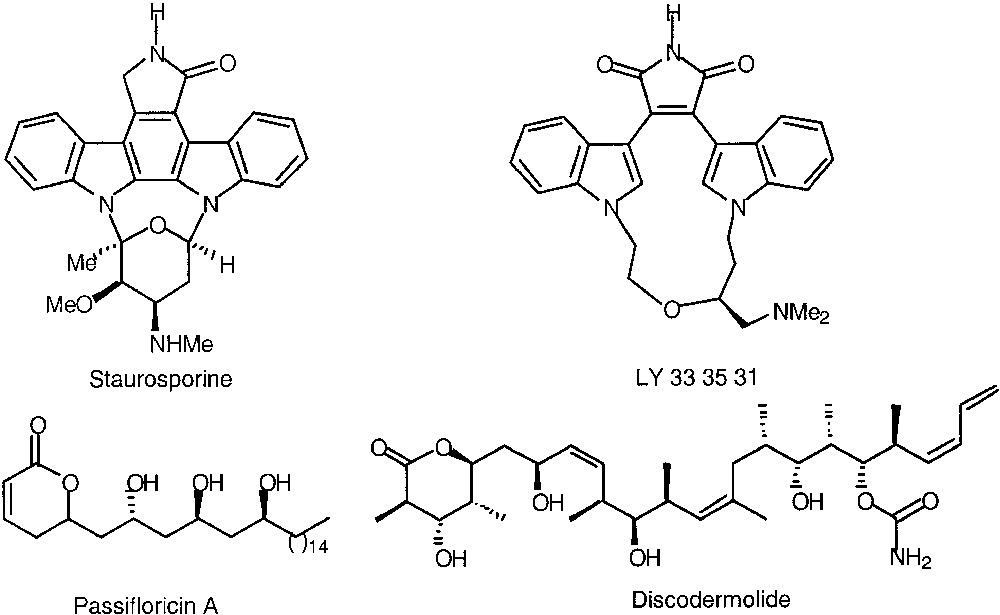
Biologically active compounds.
The first product that we became interested in was LY 33 35 31 related to the naturally occurring staurosporine, which is a Protein Kinase C inhibitor. This compound was obtained by others with alkylation of 1 with bis-mesylate 2, which was synthesized from the expensive (R)-1-chloro-2,3-propanediol 3 with an overall yield of 45–55% and an enantiomeric excess of 98% [24–26] (Fig. 3).

Retrosynthetic analysis of staurosporine.
By using an enantioselective allyltitanation, we were able to transform the cheap allylic alcohol 4 to the bis-mesylate 2 in six steps, in good yield (66%) and with good enantiomeric excess (98%) (Fig. 4) [27].

Synthesis of the precursor of LY 33 35 31.
If 1,2-diols are found in a great number of biologically active compounds, the 1,3-diol functionality is of great importance for the biological activity of many pharmaceutical products. Therefore, it is not surprising that numerous strategies have been developed for the stereoselective synthesis of compounds containing 1,3-diol units [28–30].
We have synthesized syn- as well as anti-1,3 diols of type D from aldehyde of type A with good to excellent enantiomeric excess by using the allyltitanium complexes (R,R)-I and (S,S)-I (Fig. 5). For example, 1,3-diols 14–17 were obtained in good yield (67–72%) and good enantiomeric excess (93%) from aldehyde 9 [31]. After the allyltitanation of aldehyde 9 by using (R,R)-I or (S,S)-I, the obtained homoallylic alcohols 10 and 11 were oxidatively cleaved (OsO4/NMO; NaIO4) and the unstable β-hydroxy aldehydes 12 and 13 were treated directly with the allyltitanium complexes. When the non-protected β-hydroxy aldehydes 12 and 13 were treated respectively with (R,R)-I and (S,S)-I, the syn-1,3-diols 14 and 17 were respectively obtained in high yield (79–85%) and with a diastereomeric excess superior to 95%. When β-hydroxy aldehydes 12 and 13 were treated respectively with (S,S)-I and (R,R)-I, the anti-1,3-diols 15 and 16 were isolated in high yield (79–83%) and with a diastereomeric excess superior to 95%. The formation of syn- or anti-1,3-diols from non-protected β-hydroxy aldehydes of type C by using (R,R)-I or (S,S)-I is very general. The most important feature in these allyltitanations is that 1,3-diols can be obtained without any protective or deprotective steps, which enhances the synthetic potential of this organometallic allylation reaction (Fig. 5). We have also demonstrated that acid-sensitive (silyl ether, trityl ether) or base-sensitive (benzoyl) protecting groups can be tolerated in these allyltitanations [31].

Synthesis of 1,3-diols syn and anti.
Non-protected 1,3-diols are interesting intermediates, which offer an efficient entry to the synthesis of natural products such as passifloricin A, an antifungal agent recently isolated, for which the absolute configuration of the stereogenic centres has to be confirmed by synthesis, as two different structures E and F have been reported in the literature (Fig. 6) [32].

Possible structures for passifloricin A.
We chose to synthesize structure E. The synthesis of E has been achieved from hexadecanal 18. As the reagents (R,R)-I and (S,S)-I are highly face-selective, the stereogenic centres were controlled by adding (R,R)-I and (S,S)-I to the corresponding aldehydes (Fig. 7). Compound 19 was obtained by this process and transformed into compound E in a three-step sequence. Unfortunately, the spectroscopic data do not correspond to those of passifloricin A. This leads to suspect that passifloricin A has structure F (Fig. 7).
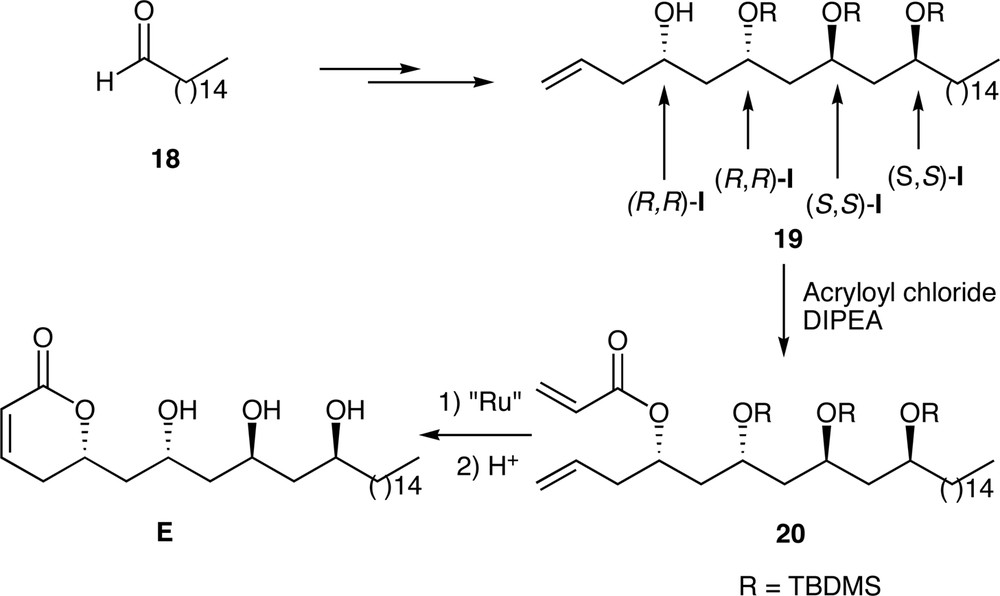
Synthesis of an analogue of passifloricin A.
The polypropionates (chains with alternating methyl-hydroxy-methyl substituents) [33] represent an important class of natural products such as the ionophores, the lactones and the macrolides [34–37]. Associated with a broad spectrum of biological activity, these structures encompass a large diversity of molecular architecture.
In looking for routes to prepare (+)-discodermolide, our interest was drawn to build up stereotetrads and stereopentads as these units are present in this compound. We have synthesized stereotetrads from meso-dialdehydes with good dia- and enantioselectivities. For example, when meso-dialdehyde 21 was treated with (R,R)-I, lactol 22 was obtained and reduced with NaBH4 to furnish compound 23 in 49% overall yield [38] (Fig. 8). It is worth noting that (R,R)-I discriminates the pro(S) face of the aldehyde. Furthermore, the allyltitanations have been shown to closely follow a Felkin–Anh transition state [38].

Synthesis of stereotetrads.
The stereotetrads have also been synthesized from aldehyde of type A with good to excellent diastereoselectivity and enantioselectivity by using the crotyltitanium complexes (R,R)-II and (S,S)-II. For example, stereotetrads 26 have been obtained from aldehyde 24 by using two crotyltitanations [39]. After treatment of aldehyde 24 with (R,R)-II, the obtained homoallylic alcohol 25 was oxidatively cleaved (OsO4/NMO, NaIO4) and the β-hydroxyaldehyde 26 was treated directly with the crotyltitanium complexes. When 26 was treated with (R,R)-II, the stereotetrads 26 and 27 were respectively obtained in high diastereoselectivity (> 95%) and enantioselectivity (Fig. 8) [39]. As in the case of the allyltitanium complexes, the crotyltitanium complexes are highly face-selective.
Stereopentads can be obtained from meso-dialdehydes by treatment with (R,R)-II and (S,S)-II. For example, when meso-dialdehydes 21 and 30 were treated with (R,R)-II, the stereopentads 29 and 32 were isolated after reduction of the intermediate lactols 28 and 31 with NaBH4 (Fig. 9) [40]. It is worth noting that compound 32 corresponds to the C15–C21 fragment of (+)-discodermolide (Fig. 10). The desymmetrization of meso-dialdehydes by using chiral titanium complexes allows a short and efficient synthesis of polypropionates.

Synthesis of stereopentads by desymmetrization of meso dialdehydes.
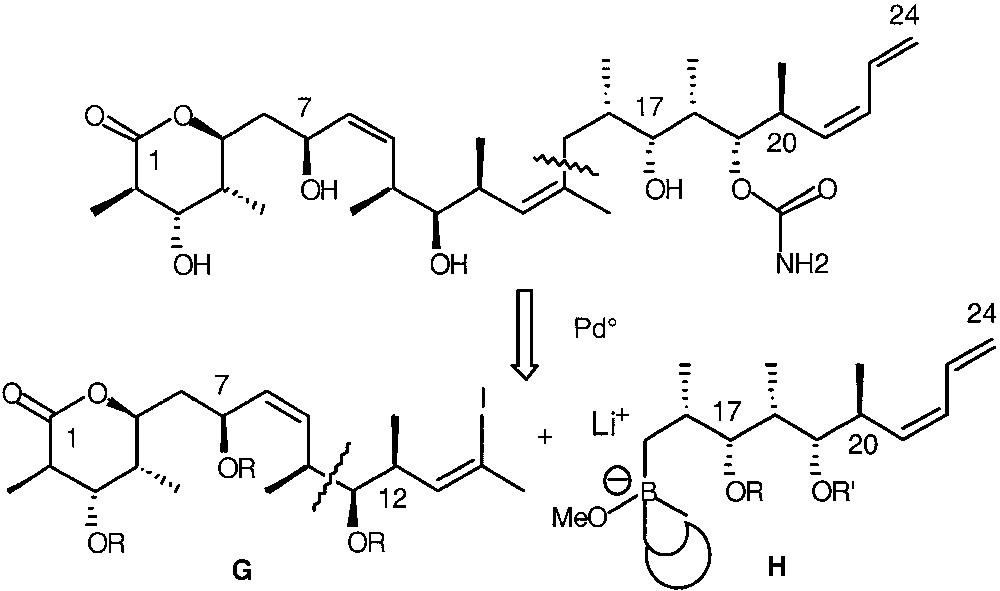
Retrosynthetic analysis of (+)-discodermolide.
As the chiral allyl- and crotyltitanium complexes can tolerate a great number of functionalities, they can be used efficiently in the synthesis of biologically active natural products. For our part, we choose to synthesize (+)-discodermolide as we have already obtained the C15–C21 fragment. (+)-Discodermolide is a unique polyketide isolated from the Caribbean deep-sea sponge Discodermia Dissoluta [41, 42]. Its structure was determined by extensive spectroscopic studies and the relative stereochemistry was assigned by single crystal X-ray crystallography. Structurally, it bears 13 stereogenic centres, a tetrasubstituted γ-lactone (C1-C5), one di- and one trisubstituted (Z)-double bond, a pendant carbamate moiety (C19) and a terminal (Z)-diene (C1-C24). (+)-Discodermolide was initially found to be a potent immunosuppressive agent, both in vivo and in vitro, as well as displaying antifungal activity. It inhibited T-cell proliferation with an IC50 of 9 nM and graft versus host disease in transplanted mice. Further biological screening of this compound revealed cytotoxicity, causing cell cycle arrest in the G2/M phase in a variety of human and murine cell lines [43]. (+)-Discodermolide has been recognized as one of the most potent tubulin polymerising agents presently known. The highly encouraging biological profile of (+)-discodermolide makes it a promising candidate for clinical development as a chemotherapeutic agent for taxol-resistant breast, ovarian and colon cancer and other multidrug-resistant cancers. Clinical development is severely hampered by the extremely scarce supply of (+)-discodermolide (0.0002% w/w frozen sponge) from the natural source. Thus, total synthesis presently provides the only viable route to useful quantities of this novel cytotoxic polyketide. Consequently, there has been considerable synthetic effort toward (+)-discodermolide.
Our approach to (+)-discodermolide is based on synthesis of two fragments G and H by using allyltitanations. Connection of G and H will then have to be carried out with the help of a palladium-catalyzed coupling reaction (Fig. 10).
The synthesis of fragment C15–C24 was achieved from meso-dialdehyde 30, which was transformed to iodide 33 in 8 steps via stereopentad 32. The synthesis of the C1–C13 fragment was achieved in 18 steps from the commercially available methyl (+)-3-hydroxy-2-methyl propionate 35, the stereogenic centre of which corresponds to C12 of (+)-discodermolide. All the stereogenic centres were controlled by using (R,R)-I (C7) and (R,R)-II (C2, C3, C4, C5, C11, C12) (Fig. 11).

Synthesis of the C1-C13 and C15-C24 fragments of (+)-discodermolide.
The transformation of 33 to the boronate 34 and the coupling of this later compound with the vinyl iodide of type G, prepared from 45, will lead to the C1-C24 fragment of (+)-discodermolide. The stereocontrolled synthesis of compounds 33 and 45 demonstrates that the highly face selective allyltitanium complexes are useful reagents for achieving remote stereocontrol in acyclic systems. Furthermore, by using these reagents, protection-deprotection sequences can be avoided as polar groups are tolerated, which increases the efficiency of the syntheses.
Acknowledgements
The COST program D13/001/00 is acknowledged for support and Eli-Lilly (Indianapolis, IN) as well as PPG-Sipsy are acknowledged for financial support.