

1 Introduction
Dendrimers as macromolecular “vehicles” for homogeneous catalysts combine a high level of structural regularity, molecular monodispersity and a wide variety of modes of attachment of well-defined catalytic sites [1–5]. A dendrimer ‘architecture’ with its iterative sequence of branching and linear growth elements [6] offers the possibility of catalyst fixation within the core (‘endodendral fixation’) or at its periphery (‘exodendral fixation’) (Fig. 1).
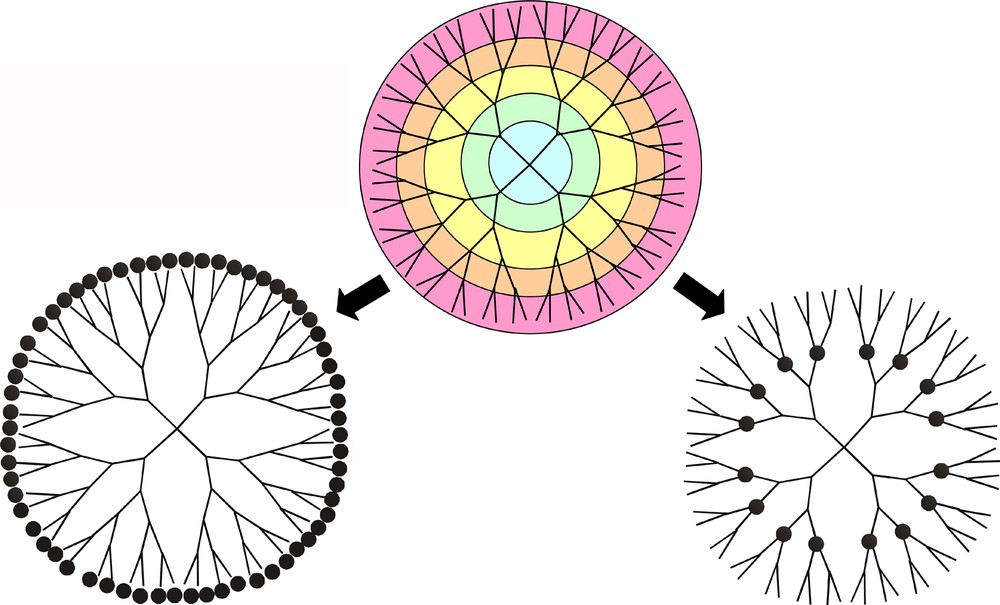
Fixation of catalytic metal centres (represented by the black spheres) in exodendral (left) and endodendral (right) positions of dendrimers
Whereas the first examples of the integration of a molecular catalyst into the core position of a dendritic structure were reported by Brunner et al., who studied the influence of a chiral dendritic periphery on the performance of asymmetric cyclopropanation catalysts [6], the fixation of catalysts at the periphery of a dendrimer was pioneered by van Koten, van Leeuwen and co-workers [7, 8]. These groups reported a study of the Kharasch reaction which was catalysed by nickel(II) complexes containing NCN-pincer ligands. Since this early work, there has been a steadily increasing number of dendrimer catalysts that have been synthesized and studied [9–11].
Catalyst recycling is an important practical objective in the development of supported molecular catalysts and, in particular, dendrimer catalysts [11, 12]. This has been achieved using membrane reactors, although frequently with deteriorating catalyst performance with time (vide infra). This utilitarian aspect of dendrimer catalysis early on provided the motivation to study chiral dendrimer catalysts which were generated either by attachment of achiral complexes to chiral dendrimer structures or the immobilisation of chiral catalysts to the established non chiral dendrimers.
Since enantioselection in a stoichiometric or catalytic reaction is governed by small increments of free enthalpy of activation [13], such transformations are in principal particularly suited to assess ‘dendrimer effects’ which result from the catalyst immobilization. In the assessment of such multi-site macromolecular catalysts it is essential to establish whether the immobilized catalyst units retain their identity and are not altered by the nature of the dendrimer backbone. The linker and spacer units employed in the fixation of the catalysts may be crucial in this respect as well as the functional groups present in the dendrimer. Regarding the dendrimer core structure itself, the length and conformational rigidity of the branches and spacers are important factors when evaluating a dendritic catalyst. For immobilized asymmetric catalysts, even subtle conformational changes may significantly influence their stereoselectivity. The interplay of all these factors will generally determine negative or beneficial dendrimer effects on catalyst performance, which will be the focus of this short overview of the field.
2 Dendrimer effects on the stability, activity and selectivity of immobilized catalysts
Whether the fixation of catalytic sites in endo- or exodendral positions enhances or inhibits the performance of a catalyst depends on the type of conversion, the choice of the dendrimer core and the mode of fixation of the catalyst. In the initial study on the dendrimer catalyzed Kharasch reaction a slight change in the regioselectivity of the reaction was observed [7]. However, as van Koten et al. showed in subsequent work, the proximity of the catalytic centres in dendrimers of higher generation inhibits the catalytic activity [14]. Another early example of a modified regioselectivity was reported by Suslik et al. for the epoxidation of olefins using manganese porphyrins carrying dendritic wedges [15].
An enhancement of the rate and selectivity in a catalytic reaction upon going from a mononuclear catalyst to a multisite dendrimer catalyst is to be expected for conversions involving bimetallic catalysis, i.e. a cooperative interaction of two catalytic centres with the substrate. The high local concentration of catalytic sites on the surface of a dendrimer may thus provide this type of bimetallic interaction. The asymmetric ring opening of epoxides catalysed by Co(salen) complexes, reported by Jacobsen et al. is known to involve bimetallic catalysis [16]. A significant increase in catalytic activity, attributed to the proximity effects between single catalyst units has been observed upon incorporation into small dendritic structures.
Reetz and co-workers prepared phosphine-functionalized poly(propyleneimine) (PPI) dendrimers and studied the catalytic activity of their Pd, Rh and Ir complexes [17]. For the Heck type C–C coupling catalysis with the Pd derivatives they found greater catalyst stability for the higher generation dendrimers than for the corresponding mononuclear system. Similar observations were made by Caminade, Majoral and their co-workers in a study of the ruthenium-dendrimer catalyzed Knoevenagel reaction [18]. Positive dendrimer effects have also been observed in several other recent studies [19].
The aspect of catalyst activity and stability is of great importance in the application of dendrimer catalysts in continuous reactors. Palladium-catalyzed reactions have received most attention in this context [20–22]. In general, the behaviour of dendrimers in continuous flow systems was found to be in contrast to their frequently high stability in batch reactions. Small changes in the dendrimer structure may induce large variations in catalyst stability as van Leeuwen et al. have shown for Pd-catalyzed allylic substitutions employing functionalized carbosilane dendrimers [20b]. When dendrimeric ligands with Si(CH2PPh2)2 end groups were used, the yields for allylic substitution reactions dropped rapidly during continuous experiments, however, when SiCH2CH2PPh2 end groups were used, stable catalysts were obtained (Fig. 2).

Pd-catalyzed allylic substitutions employing functionalized carbosilane dendrimers: synthesis of the phosphine dendrimer (top) and catalytic activity in a continuous-flow reactor (bottom) [20b].
Catalyst deactivation when using a first generation phosphine-ester functionalized carbosilane dendrimer was also observed by van Koten, Vogt et al. [21, 22]. In the Pd-catalysed hydrovinylation of styrene, which they performed in a continuous-flow reactor, precipitation of Pd metal in the reactor and on the surface of the membrane took place during the course of the reaction. This deactivation of the dendrimer catalysts was attributed to the formation of multiply coordinated phosphine complexes and multinuclear phosphine-bridged complexes. Their formation is possibly a result of the high local concentration of phosphine in the periphery of the dendrimer.
3 Dendrimer fixation of chiral catalysts
The underlying concept of Brunner's ‘dendrizymes’ (vide supra) [6] was the creation of a chiral dendritic architecture in the environment of a catalytic centre which should induce stereoselectivity in the same way as in enzymes. In metalloenzymes, in particular, the first coordination sphere of the metal centre is frequently achiral (see for instance the ubiquitous tris-histidine binding sites!) and the chiral induction is due to the chirality of the polypeptide protein structure, which allows for chiral secondary interactions with the substrates. In contrast to the biological systems, the reaction performance of dendrimers containing a chiral dendritic backbone has been found to be unsatisfactory [23]. This may be due to the fact that the dendrimers, which were employed, do not form well-defined secondary structures as polypeptides do. Consequently, the arrangement of functional groups in the ‘second sphere’ of the catalytic site and thus their interaction with a potential substrate remains ill defined.
In view of the inefficiency of the chiral induction effected by chiral dendritic structures, most attention has been directed towards the covalent or electrostatic [24] fixation of established chiral mononuclear complexes to achiral dendritic cores or wedges. A number of efficient chiral dendrimer catalysts have been obtained as will be discussed in the following sections. However, even in these cases particular care has to be taken to avoid the negative interference of functional groups in the dendrimer core with the attached catalytic sites. This aspect is thought to be important in the enantioselective ethylation of benzaldehyde with chiral dendrimer catalysts, for which both the chemical yields and the enantiomeric excess were found to decrease with increasing generation of the dendrimers [23]. Multiple interactions of the catalytic sites on the dendritic surface with the substrate at higher generations add to the observed negative effects. To relieve these interactions with the dendrimer end groups at the periphery of higher generations, the introduction of an alkyl chain as a spacer or the use of a rigid hydrocarbon backbone have been proposed [25].
3.1 Asymmetric catalysis with polycationic rhododendrimers
The first example of asymmetric rhodium catalyzed hydrogenation of prochiral olefins was reported by Togni et al., who immobilized their chiral ferrocenyl diphospines (‘Josiphos’) at the endgroups of dendrimers, thus obtaining systems of up to 24 chiral metal centres in the periphery (Fig. 3) [26–28]. The fact that the catalytic properties of the dendrimer catalysts were almost identical to those of the mononuclear catalysts was interpreted as manifestation of the independence of the individual catalytic sites in the macromolecular systems.
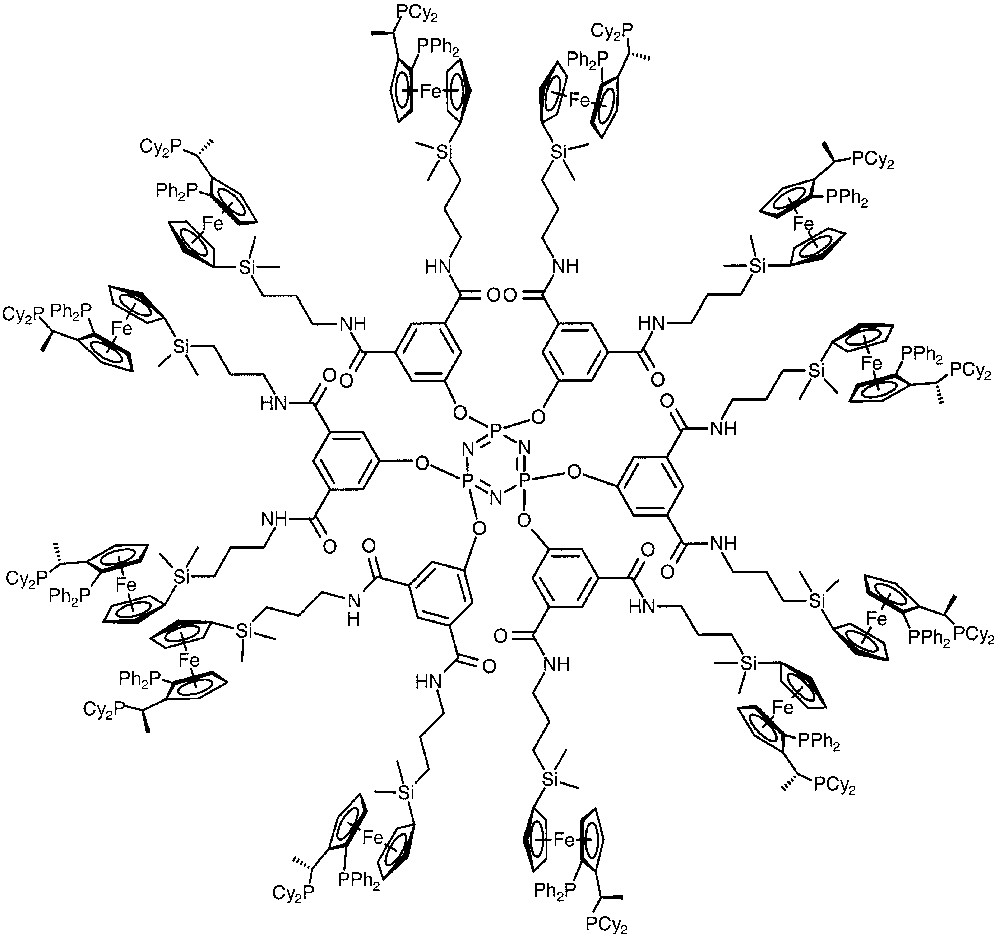
Fixation of the chiral ‘Josiphos’ ligands to a first-generation dendrimer containing a cyclotriphosphazine core.
In a comprehensive study carried out by the same group, the asymmetric hydrogenation of dimethyl itaconate, asymmetric allylic substitution and asymmetric hydroboration reactions catalyzed by the multi-Josiphos rhododendrimers were investigated [29]. The stereoselectivities obtained with the monodisperse dendrimer catalysts with up to 16 metal centres in the periphery were found to be very similar to those of the mononuclear system of reference. The authors concluded that the absence of negative dendrimer effects is probably the best possible result when cooperativity effects between single catalyst units do not play any relevant role. Small losses in selectivity, observed upon going to higher dendrimer generations, may be due to local concentration effects that become important, in particular for cationic catalyst species.
A series of chiral phosphine-functionalized poly(propyleneimine) (PPI) dendrimers was synthesized by reaction of carboxyl-linked C2-chiral pyrphos ligand (pyrphos = 3,4-bis(diphenylphosphino)pyrrolidine) with 0th-4th generation PPI using ethyl-N,N-dimethylaminopropylcarbodiimide (EDC)/1-hydroxybenzotriazole as a coupling reagent [30]. The dendrimers obtained were characterized by NMR spectroscopy, elemental analysis as well as FAB and MALDI-TOF mass spectrometry establishing their molecular masses of up to 20 700 amu. Metallation of the multi-site phosphines with [Rh(COD)2]BF4 cleanly yielded the cationic rhododendrimers containing up to 32 metal centres (for the 4th generation species) (Fig. 4). The complete metallation of the chiral phosphine sites was demonstrated by 31P NMR spectroscopy.

Synthesis of the metallodendrimers by reaction of the pyrphos-functionalized PPI dendrimers of the 0th–4th generation with [Rh(COD)2]BF4.
The relationship between the size/generation of the dendrimer and its catalytic properties was established in the asymmetric hydrogenation of Z-methyl-α-acetamidocinammate and dimethyl itaconate. A decrease in both activity and selectivity of the dendrimer catalysts was observed on going to the higher generations (Fig. 5). An important factor governing the properties of the metallodendrimers as catalysts is certainly the flexibility of the dendrimer core which is high in the given case and allows for the bending of the attached rhodium complexes back to the inside of the otherwise globular molecule. This reduces the accessibility of the catalytic centres and at the same time renders their immediate environment less uniform than originally envisaged. A more rigid dendrimer core such as the polyamide core of the family of PAMAM dendrimers may partially suppress this negative effect.
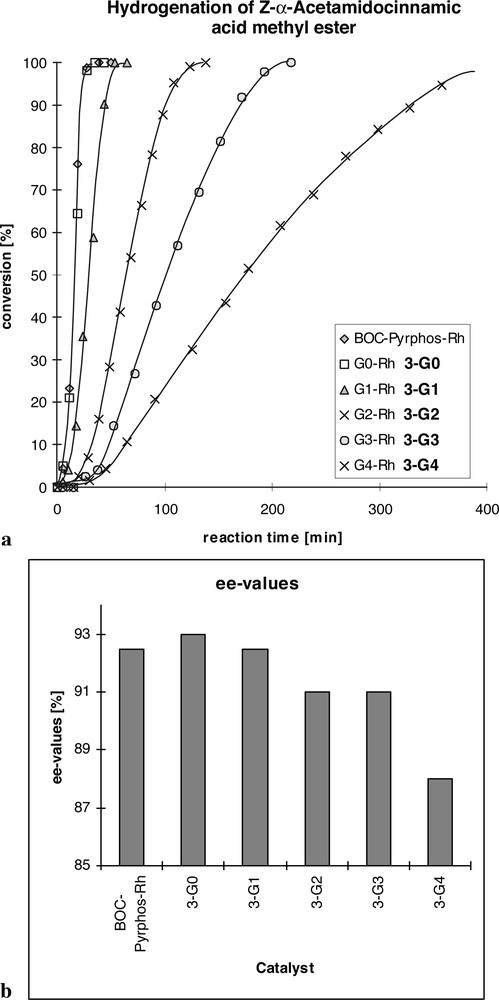
(a) Conversion curves of the asymmetric hydrogenation of Z-methyl-α-acetamidocinnamate for the different catalyst generations; (b) enantioselectivity for the different catalyst generations.
3.2 Strongly positive selectivity effects in high-generation dendrimers bearing chiral phosphine–palladium complexes
In an effort to extend the use of the pyrphos-derived dendrimers to asymmetric Pd-catalyzed coupling reactions, strongly positive selectivity effects were observed upon going to very large multisite chiral dendrimer catalysts. This enhancement of the catalyst selectivity was observed in palladium catalyzed allylic substitutions that are known to be particularly sensitive to small changes in the chemical environment of the active catalyst sites [31].
The chiral pyrphos-functionalized dendrimers, employed in this study, were both the PPI derivatives mentioned above (containing 4, 8, 16, 32 and 64 metal binding sites) and the analogous poly(amidoamine) (PAMAM) dendrimers (containing 4–64 binding sites) which were synthesized in an analogous way by ethyl-N,N-dimethylaminopropylcarbodiimide (EDC) induced amide coupling of the ligand-linker unit 1 with the amino end groups of the dendrimer (Scheme 1) [32].

Synthesis of the Pyrphos-functionalized PPI and PAMAN dendrimers employed in the asymmetric allylic amination of 1,3-diphenyl-1-acetatopropene.
The metallation with appropriate Pd-complexes gave the corresponding catalyst precursors. The allylic amination of 1,3-diphenyl-1-acetatopropene with morpholine was the test reaction for probing the catalytic performance (Eq. [1]).
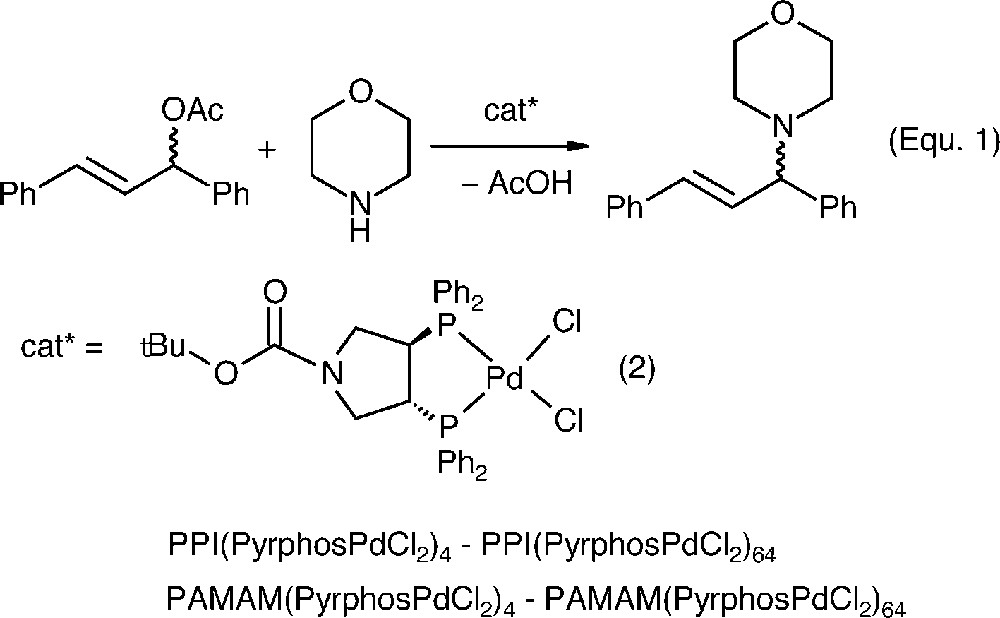
The mononuclear catalyst [(Boc-pyrphos)PdCl2] (2), which is very unselective for this transformation (9% ee) provided the point of reference for the subsequent studies with the dendrimer catalysts. Using complex 2 or the metalladendrimers PPI(PyrphosPdCl2)4–PPI(PyrphosPdCl2)64 and PAMAM(PyrphosPdCl2)4–PAMAM(PyrphosPdCl2)64 in 0.3 mol% effective catalyst concentration, the amination of 1,3-diphenyl-1-acetatopropene gave the results displayed in Fig. 6.
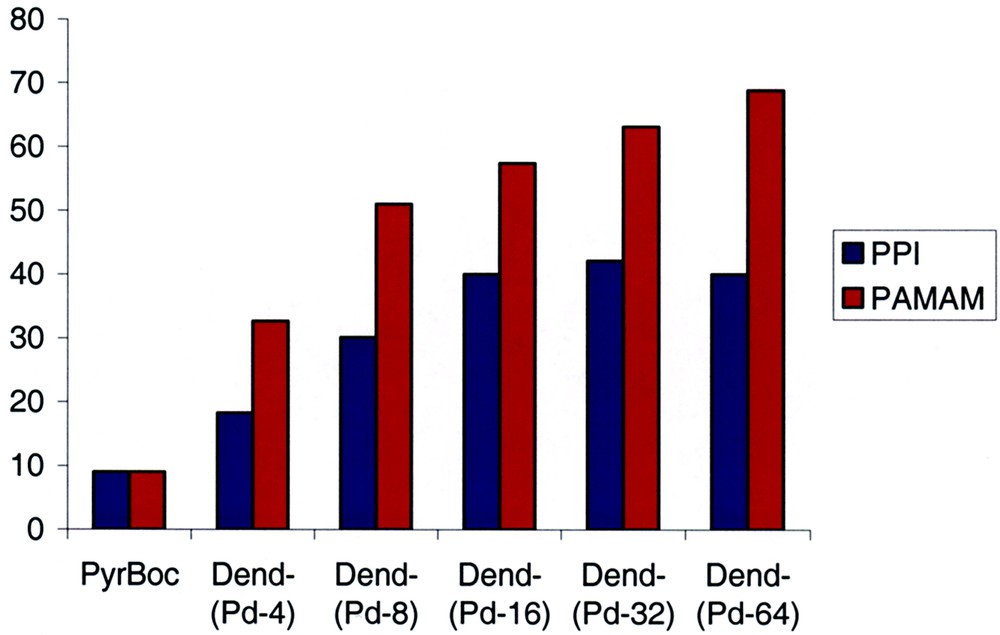
Dependence of the enantiomeric excesses found for the reaction in Eq. (1) on the dendrimer generation for both precatalyst series PPI(PyrphosPdCl2)4–PPI(PyrphosPdCl2)64 and PAMAM(PyrphosPdCl2)4–PAMAM(PyrphosPdCl2)64.
A remarkable increase in catalyst selectivity is observed as a function of the dendrimer generation. This steady increase in ee-values for the allylic amination is less pronounced for the PPI-derived catalysts [40% ee for PPI(PyrphosPdCl2)64] than for the palladium-PAMAM dendrimer catalysts for which an increase in selectivity from 9% ee for 2 to 69% ee for PAMAM(PyrphosPdCl2)64 was found. The same general trend was observed in the asymmetric allylic alkylation of 1,3-diphenyl-1-acetatopropene with sodium dimethylmalonate, which indicates that the results of the amination reaction may be typical for allylic substitutions in general [32]. The underlying mechanistic reasons for this remarkable ‘dendrimer effect’ remain to be elucidated. However, it is well known that for C2-chiral Ph2P–diphosphines the orientation of the phenyl substituents controls the stereoselectivity in the catalytic transformations [33]. Upon going to the higher dendrimer generations with their increasingly crowded diphosphine periphery, the conformational flexibility and preferential orientation of these aryl substituents may be altered which in turn changes their selectivity in the allylic substitutions.
3.3 Asymmetric catalysis in the core of dendrimers
Brunner’s concept of attaching dendritic wedges to a catalytically active metal complex represented the first example of asymmetric catalysis with metal complex fragments located at the core of a dendrimer [6]. Important early examples of catalysts in core positions were Seebach’s Taddol systems [34, 35]. In general, the catalytic performance of such systems was either unchanged with respect to the simple reference or significantly lower. In no case the potential analogy between this core fixation and the existence of efficient reactive pockets in enzymes has been vindicated. As already discussed above, this may be due to the absence of defined secondary structures in the dendrimers that have been employed to date.
Two interesting recent reports of dendritic core-functionalized Ru-BINAP catalysts, which were employed in asymmetric hydrogenations and which were fully recoverable, have appeared recently [36, 37]. In particular, such dendrimers containing long alkyl chains in the periphery were synthesized and employed for asymmetric hydrogenation using a mixture of ethanol/hexane as reaction medium (Fig. 7).
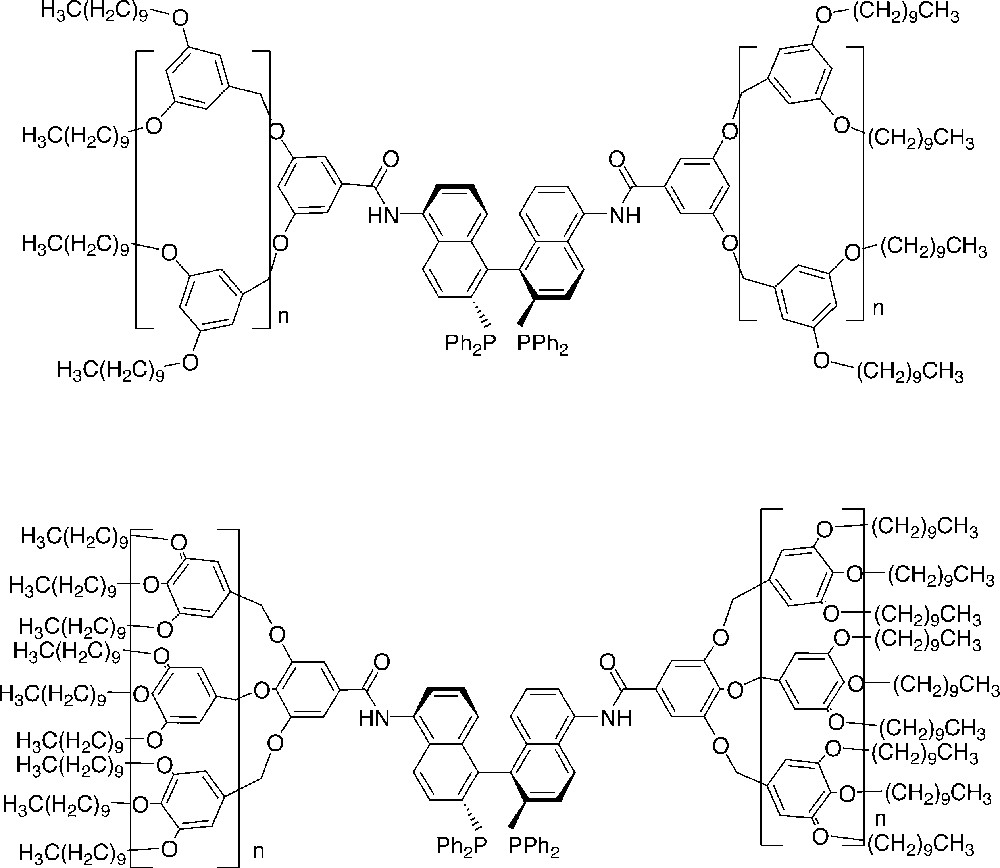
Ru-BINAP core-functionalized dendrimers containing long alkyl chains in the periphery which were employed for asymmetric hydrogenation using a mixture of ethanol/hexane as reaction medium.
Unlike the thermomorphic systems, this binary solvent system provided complete miscibility of the phase over a broad range of reaction temperatures and avoids the use of water during the catalytic conversion with its established negative effects on the enantioselectivity [37]. Phase separation after complete reaction was induced by the addition of small quantities of water and the recycling of the catalyst thus readily achieved. Remarkably, the dendrimer-attachment to the BINAP system did not lead to a decrease in selectivity.
4 Conclusion and outlook
As emphasized in this short overview, the stereoselection in asymmetric catalytic transformations depends on very small energy increments. It is therefore an excellent sensitive probe for ‘dendrimer effects’ and will continue to be studied in this fundamental context. However, the results obtained in recent years have also established that the structural characteristics of the established dendrimer systems, such as the absence of a well defined secondary structure, has limited the development of efficient abiotic enzyme mimics based on dendrimers. To achieve this ambitious goal, more efforts in dendrimer synthesis will be necessary.
Chiral dendrimer catalysts have been successfully employed in continuous-flow membrane reactors. As monodipersed macromolecular supports they provide ideal model systems for less regularly structured but commercially more viable supports such as hyperbranched polymers. The use of dendritic catalysts in biphasic solvent systems has only just begun and appears to be a particularly fruitful field for further developments. These utilitarian aspects aside, it is the aesthetic attraction of these topologically highly regular macromolecules that continues to fascinate those working in the field of dendrimer catalysis.