

1 Introduction
Everywhere in nature branched or dendritic architectures can be found. Not all of them are really perfect and e.g. the attractivity of a tree or a full forest is given only by a certain irregularity and definitely a high versatility. But what about chemistry? Do we need to be perfect? Or is a perfect synthetic material possible at all? It is obvious that, when posing the question like this, one has to answer with no. However, when one enters the field of dendritic molecules in polymer science and the question arises whether dendrimers are better than hyperbranched polymers no easy answer can be given. First let us clarify the terms: dendritic means highly branched tree-like structures and covers both, dendrimers as well as hyperbranched polymers but also other highly branched or fractal molecules; dendrimers are perfectly branched molecules prepared in a step-wise manner with the potential to come close to structural and molar mass uniformity; on the other hand hyperbranched polymers are prepared in a random one-pot synthesis from monomers having branching potential (e.g., AB2 based) but with low control over structure and molar mass.
After about 15 years of intensive research in this field and a giant number of publications – over 5000 publications appeared between 1996 and 2001 (taken from [1]) – there are still surprises on both sides and still open questions to be answered. Instead of having a clear pictures by now one has to state that dendrimers as well as hyperbranched polymers both offer a big chance but also an ultimate challenge in the material science without giving preferences to any side. Dendrimers are so attractive because of their perfect structure and exact molar mass and all the possibilities to mimic very complex molecules as found in nature, but at the same time the synthesis of high generation dendrimers without any defects can be considered one of the major challenges in organic chemistry. The synthesis of hyperbranched polymers, on the other side, might not be trivial but certainly feasible. But the full characterization of the highly complex and irregular structure and the establishment of a clear structure property relationship remains a challenge since a hyperbranched polymer consists of a huge amount of different isomeric macromolecules besides its large polydispersity.
Of course, huge progress has been made in all directions regarding synthesis, characterization, theory, and modelling, and also with regard of potential applications. Several dendrimers and hyperbranched polymers have been commercialised e.g. by Dendritech®, Inc. (Starburst®), Qiagen (SuperFect®), DSM (Astramol, HybraneTM) and Perstop (Boltorn®) and the variety in the synthesized structures is very impressive as well as the intensive research activities on characterization and property determination as summarized in a large number of reviews (for some more recent reviews, see [1–11]). Out of this broad range of results I would like to focus purely on hyperbranched polymers predominantly from our own work and to present a few examples on the synthetic side with structure analysis as well as some results how the end group functionality of the hyperbranched polymers strongly determines the property profile and can be utilized to tune the properties. Hopefully one will see that the chances and the challenges hyperbranched polymers present are well taken care of.
2 Polymer synthesis and structural analysis
Hyperbranched polymers are synthesized classically by polyaddition or polycondensation reactions of ABx monomers as described early on by Flory [12] (Fig. 1) having dendritic (D), terminal (T) and linear (L) units as well as ideally one focal group. More recently, also other reactions including self condensing vinyl polymerization (SCVP) [13–20] and ring-opening multibranching (ROMBP) polymerisations [21–26], and also metal catalyzed polymerization reactions (see: ‘chain walking’) [27–31] have been used to prepare highly branched macromolecules which were also termed ‘hyperbranched’. These newer approaches usually cannot lead to random or statistical hyperbranched polymers with a degree of branching of 50% (D+T units/D+T+L units) due to a different reactivity of the B functions in the monomer. This deviates from the original prerequisites posed by Flory, which imply also the need for reaction only between A and B without any side and cyclisation reactions. Personally, I tend to consider only those polymers as ‘hyperbranched’, where each repeating units presents the possibility for branching. By this definition, polyethylene by the ‘chain-walking’ mechanism, as well as the arborescent graft and CombburstTM polymers [32–40] do not belong to this category, since all these structures contain oligomeric or polymeric chains of monomers without branching option between the branching points.
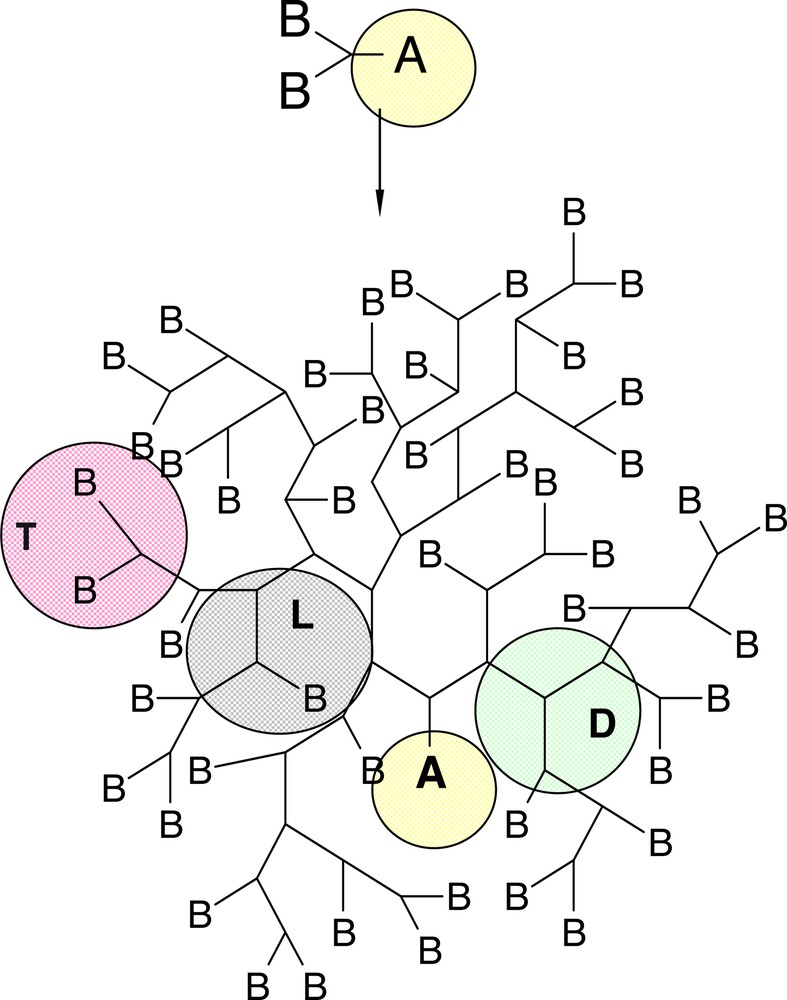
Schematic representation of a hyperbranched polymer by AB2 monomers exhibiting linear (L), dendritic (D) and terminal (T) units as well as one unreacted A functionality as focal unit.
But even when the focus is laid on the ‘classical’ hyperbranched polymers, where ABx (mostly AB2, equal reactivity of all Bs) or ABB′ (different reactivity of Bs) monomers with or without a core molecules (Bx) are used, the variety of reported structures is huge and can no longer be reviewed completely. Certainly the majority is prepared by polycondensation as hyperbranched polyesters, polyamides, polyethers, polyesteramides, polysulfones, polyetherketones, polyphenylenes, etc., but increasingly also by polyaddition leading to e.g. polycarbosilanes, polyurethanes, polyarylenes, polyetheramides or polythioethers (see [3–9] and references herein). Thus, the hyperbranched polyetheramide synthesis developed in our own group [41] used the ring opening addition reaction of phenols towards oxazoline units (Fig. 2) and resulted a very well soluble, amorphous product with a low solution viscosity.
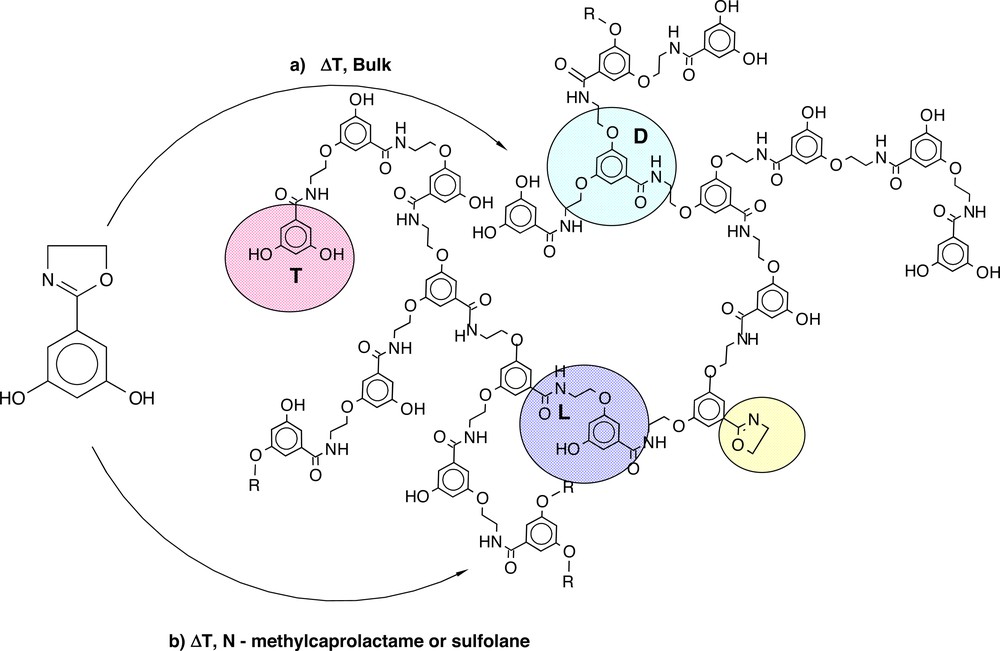
Hyperbranched polyetheramide by the ring-opening addition reaction of phenols to oxazoline [41].
Especially cycloaddition reactions offer the advantage of an often very selective and clean high yield reaction not influenced by special functionalities. The relatively easy synthesis of the hyperbranched polyphenylenes by Müllen et al. [42] is an excellent example for this. In addition, certain cycloaddition reactions form as linear units non-stable intermediates, which allow the preparation of hyperbranched polymers without any linear units [43,44] and therefore exhibiting formally a degree of branching (DB) of 100% (Fig. 3). Similarly, recently Smet et al. [45] used the acid catalyzed condensation of isatin with aromatic compounds for the synthesis of hyperbranched polyaryleneoxindoles (Fig. 4) using the fact that isatin exclusively reacts to 3,3-diaryloxindoles and therefore the linear units can be excluded from the structure. Hobson and Feast [46] described even a simple one-pot route towards nearly perfect poly(amidoamine) dendrimers by the Michael addition reaction.

Hyperbranched polymer without linear units by criss-cross-cycloaddition reaction [44].

Hyperbranched polyaryleneoxindoles without linear units (adopted from [45]).
Whereas special addition and condensation reactions allow to even avoid linear units the SVCP and ROMBP (ABB′ monomers) fight with the problem that the reactivity of both B functions are certainly not equal reducing DB. However, when one looks closely into all hyperbranched structures and when an intensive elucidation of the different structural units is achieved, only a very small number of reactions qualifies as a ‘ideal’ statistical hyperbranched reaction following the rules of Flory. Even in the plain AB2 polycondensation of 3,5-[bis(trimethylsilyloxy)]benzoylchloride we found that DB approaches usually about 60% due to a slight activation of the reactivity of the phenolate intermediate when the phenol group in meta position is already esterified [47,48]. The relative reactivity constants of the different reaction pathways could even be modelled numerically (Fig. 5) after following the development of the different structural units with conversion by detailed NMR analysis [47,48]. In contrast, the polycondensation of 4,4-bis(4′-hydroxyphenyl)pentanoic acid is strictly statistical due to separation of the reacting groups and obviously no steric hindrance [49]. In addition, side reactions can be numerous with cyclisation of the focal units as one of the most prominent example [50]. Intramolecular cycles usually limit only the achievable molar mass, whereas other non-desired reactions like ether formation during the polycondensation of bis(hydroxymethyl)propionic acid, lead to intermolecular reactions and finally to crosslinking [51].

Different reactions pathways in the polycondensation of 3,5-[bis(trimethylsilyloxy)] benzoylchloride and the matrix notation for the twelve rate constants; for ideal random branching all reaction constants are equal (Kideal-matrix) but for this aromatic AB2 monomer differences could be identified by modelling numerically the evolution of the structural features (see Kfit-matrix) [47,48].
The picture becomes even more complicated when an alternative synthetic route enters the discussion of hyperbranched polymers: the reaction of A2 monomers with B3 monomer. This is old chemistry going back to network formation [12], but the resulting products still qualify as a hyperbranched polymers before the gel point is reached and the gel point can be avoided using the ideal ratio A to B (optimum seems to be 2:3 with the aim of reasonable molar masses) and optimised reaction conditions [52–56]. The number of different repeating units, however, increases. In our kinetic evaluation of hyperbranched polyesters from AB2 monomers, we determined 6 different repeating units (Fig. 6) [49] depending whether the focal unit was still present (e.g. the first structural unit would be the monomer). In a product formed e.g. by p-diaminophenylene + trimesic acid (Fig. 7) [52] even 10 different structural units have to be considered focusing on monomer B3, six of them where found in the reaction product by NMR analysis (Fig. 8, marked area) [57]. A major difference of the A2+B3 products from the classical approach is that depending on the ratio A to B functions several functions A might be present in the hyperbranched molecule which finally will cause the crosslinking (Fig. 7).
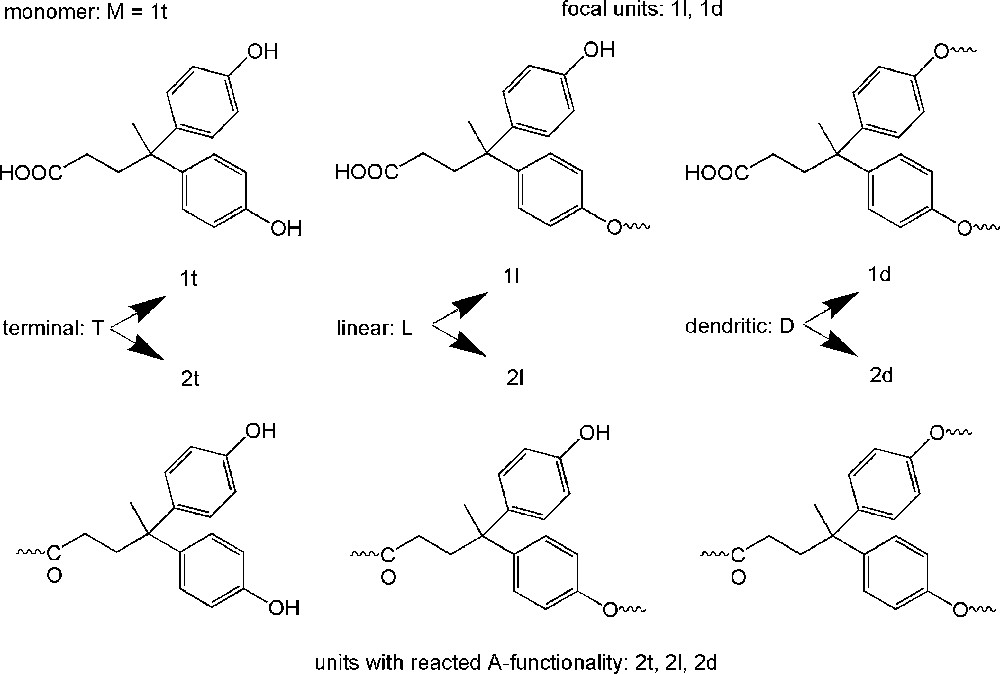
Six different repeating units starting from the AB2 monomer 4,4-bis(4′-hydroxyphenyl)pentanoic acid which have to be considered when the development of the DB with conversion is followed [49].

Reaction of trimesic acid and p-phenylene diamine and the resulting percentage of the different structural units depending on the functionality ratio in the feed [57].
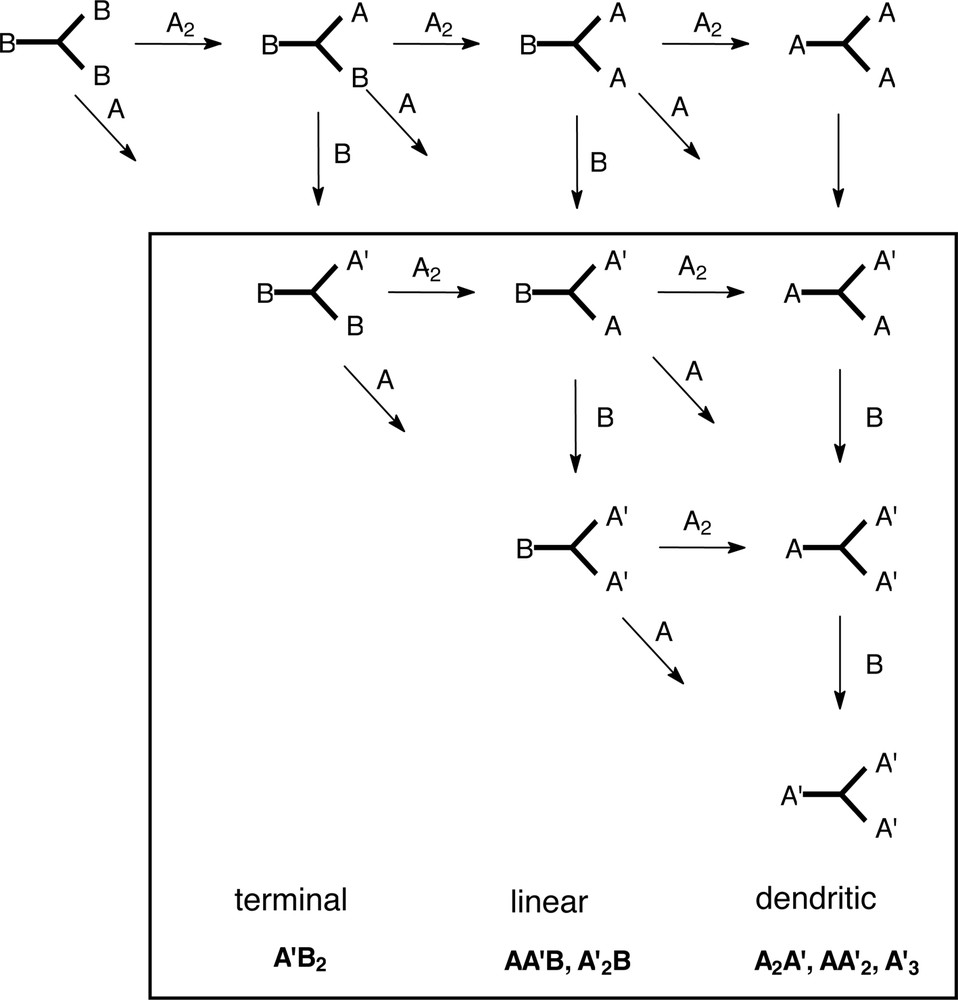
Ten different repeating units which have to be considered in the polycondensation of trimesic acid and p-phenylene diamine (see Fig. 8); the units in the frame could be identified in the hyperbranched product by NMR analysis [57].
There is the possibility to improve the A2+B3 approach by using monomers with a selectively higher reactivity of one A (AA*+B3) or even an A and an B function (AA*+B*B2) favouring the formation of an A(A*–B*)B2 intermediate. Several examples have been reported already in the literature in this regard [58–62]. One prominent example is the formation of hyperbranched poly(urea-urethane)s as reported by Bruchmann et al. [63–65] and also very recently by Yan et al. [66]. Here, the final structure contains both urea and urethane groups and both exist in linear, terminal and dendritic units (Fig. 9). Nevertheless, even in this very complicated structure NMR proved to be a very powerful tool and a complete structure analysis was possible with verifying a DB significantly above 50% (between 60 to 70%) [67].

Hyperbranched polyureaurethane by the AA*+B*B2 approach [64,67]
Besides the complex structure, also molar mass determination for hyperbranched polymers is far from trivial. By theory [12] the molar mass distribution of ‘classical’ hyperbranched polymers reaches infinity, which means in reality that quite broad molar mass distributions are faced, which cause problems applying light scattering techniques or MALDI–TOF. In the first method, Mw values might be determined quite reasonably but no structure factor can be obtained due to the broad molar mass distribution. The second method, MALDI–TOF, provides information on the repeating units and even on side reactions but one cannot assume that the exact molar mass or molar mass distribution is given since branched molecules of different molar mass might desorb from the matrix with a different probability. It is obvious that molar mass determination by GPC lacks the fact that linear standards are not suitable for calibration. The application of light scattering and viscosity detector in the GPC as mostly done for branched polymers improves the results but still the broad molar mass distribution and the large number of the polar end groups might cause problems. Recently we were able to calibrate GPC data with perfectly branched dendrimers and to perform a preparative fractionation of hyperbranched polyester and polyetheramide samples [68–70]. Up to 20 fractions some having Mw/Mn values below 1.3 (Fig. 10) could be obtained and analysed which allow now a more precise structure and molar mass determination of the single fractions with the option to obtain information on the shape of the molecules form light scattering and to follow shape changes with molar mass increase finally allowing to verify GPC molar mass data. The availability of fractions of hyperbranched polymers having a narrow molar mass distribution can be considered a major step forward in order to compare the molecular shape and finally also the properties of hyperbranched polymers with those of dendrimers.
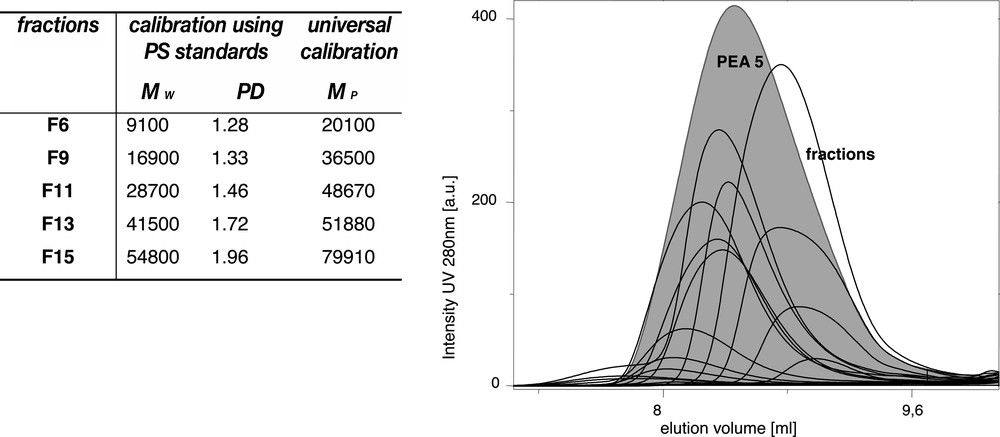
Results of the preparative fractionation of a hyperbranched polyetheramide sample PEA5 (compare with Fig. 2); GPC results on the molar masses of the fractions deviate depending on the calibration method [69,70].
3 Property profile
When asked what are the special features of hyperbranched polymers without any reluctance the better solubility and lower solution viscosity compared to linear analogues due to the branched and more dense molecule structure are highlighted. In general, this statement is certainly correct and these properties in combination with the more easy one-pot synthesis were also the reason for a relatively high interest in industry on hyperbranched polymers. However, after intensive studies and a large amount of information now available on hyperbranched polymers, the answer regarding a full property profile of hyperbranched polymers is no longer that easy. The final properties of hyperbranched polymers are determined firstly, by the structure of the repeating unit, and secondly, by the nature of the resulting end groups, or even vice-versa. Only when those two effects are clarified, one can look for the effect of the degree of branching, which is even further overlapped by the molar mass and the molar mass distribution influence. The major problem to fully understand the final effect of the branching is the lack of appropriate linear analogues for comparison. Of course, a hyperbranched polymers having a huge number of functional and/or polar end groups must have a different solubility behaviour than a linear polymer only having maximum of two end groups at the chain end. In addition, linear polycondensates might have a strong tendency for crystallization whereas hyperbranched polymers are usually fully amorphous which also influences strongly solubility as well as viscosity. Therefore, a comparison is only possible when the linear model and the hyperbranched polymers have the same type and number of (functional) end groups! Luckily, there is at least one study in the literature [71] on linear and dendritic polyesters where this direct comparison is given. The authors demonstrated that the thermal transitions as the glass transition temperature (Tg) are identical independent of the architecture and only dependent on the nature of end functionality. On the other hand, the solubility is definitely increased by the branched structure and a further enhancement in solubility was found when one switches from hyperbranched to perfectly dendritic molecules.
The advantage of the low viscosity of hyperbranched polymers also has to be discussed carefully: mostly, the solution viscosity of hyperbranched polymers is analysed, and again compared with that of non-functional linear analogues. In solution, hyperbranched molecules have to adopt a more densely packed structure due to the high branching, however, the functionality influence cannot be neglected. Therefore, the error in GPC analysis of hyperbranched polymers with RI detectors and linear standards for calibration is strongly dependent on the choice of the standard. Hyperbranched polyetheramide having polar phenolic end groups exhibits good interactions with the applied eluent DMAc/LiCl/water and in the frame of the possibility given by the branching, the molecules are relatively expanded. We found that depending whether linear polystyrene, polyvinylpyridine or PEO is used as calibration standards, the molar mass of the hyperbranched polymer as determined by scattering methods was approached better or worse [68–70]. Clearly, the interaction with the solvent, which is strongly governed by the large number of polar end groups, influences the hydrodynamic radius and the solution viscosity. There are also indications that the structure of a low molar mass hyperbranched polymer cannot be considered very dense and compact but resembles more an open fractal with even relatively large molecule dimension when the repeating units are stiff. Here, the influence of the end groups is even stronger and solution viscosity and hydrodynamic radius might even exceed those of linear (even functional) analogues, leading for instance to the determination of too high molar masses in GPC. In the fractionation experiments on hyperbranched polymers described above [69,70] we have first indications that within one hyperbranched polymer samples there is a mixture of molecules with a dependence of their shape on the molar mass as observed also previously for dendrimers [72,73]. This leads finally to the fact that there will never be an easy linear correlation of any calibration curve, which is suitable for hyperbranched polymers with a broad molar mass distribution. As discussed previously by Kim et al. [8], hyperbranched polymers consist of a huge amount of isomers for a given molar, which differ in the internal structure due to the random branching, and in addition they differ in molar mass, in shape, in DB, and maybe even in structure due to the presence of side reactions like cyclisation in low molar mass fractions, all within one polymer sample.
Of course, similar to the solution properties, the bulk properties are also determined by all the different discussed factors. The strong dependence of the glass transition temperature on the end groups in hyperbranched polymers is often stated. For instance, the Tg of an aromatic polyester can vary between –50 to +250 °C, depending whether a long alkyl chain or a polar COOH groups is introduced as functionality [74,75]. The effects are similar for other hyperbranched molecules [76–78] and it is obvious that in this case the glass transition does not represent the flexibility and the free volume of the repeating unit alone but also the strong interactions especially of polar end groups. This leads also to a very peculiar melt viscosity behaviour of hyperbranched polymers. Despite the branched structure, hyperbranched polymers with polar end-groups show in melt viscosity an elastic behaviour with the complex viscosity exhibiting a strong dependence on the applied frequency (decrease with increasing frequency) [75,79–83]. Thus, at low frequency, the polar hyperbranched polymer might readily exceed the melt viscosity of even high molar mass but viscous linear polymers. As soon as the polar end groups are modified with non-polar functions, the sample becomes predominately viscous and melt viscosity is drastically reduced (Fig. 11). In interactions of hyperbranched polymers with linear polymers, e.g. in blends or when used as additives, there is no possibility up to now to fully predict the effect on the melt viscosity. Both has been found up to now: increase and decrease of the melt viscosity of the linear matrix, dependent not only on the functionality of the hyperbranched molecules but the molar mass and molar mass distribution of the HBP, the used amount, as well as the mixing temperature and procedure [75, 79–84].

Melt viscosity in dependence on the applied frequency of hyperbranched polyetheramide (PEA) having OH end groups, modified with C12 alkyl chains and for comparison of linear polyamide 6 (PA6) [79–81].
Even though it is difficult to evaluate the property profile of hyperbranched polymers completely and to correlate it solely to the highly branched structure due to overlapping influences of different structural parameters, there is no doubt on their interesting properties for special applications. The combination of the highly branched structure and the high functionality in a constraint environment favour their use in coatings and resins [85–88], but also in thin functional films e.g. for sensorics [89–92]. Furthermore, end group modification allows ideally to optimise their properties for special applications and to fine tune e.g. miscibility, melt rheology, surface and optical properties as well as biocompatibility [3–11,89–92]. Thus beside the classical field of coatings and blends [3–9,93–95] applications in nanoscience, microelectronics, information technology, optics, and medicine are no longer out of the range of hyperbranched polymers [2–11,96,97].
4 Conclusions
At the moment, it looks like there is still no limit and no end for further studies on hyperbranched polymers. Without any question, there has been large progress in synthetic approaches, structural analysis and understanding of the properties but there are also still many open questions. Furthermore, when the full potential of organic chemistry is used, even new and more beautiful structures can be designed, synthesis can be further simplified and structural parameters will be better controlled. These new structures might on the one hand lead to new applications but on the other hand might just allow a better understanding of the potential of these highly branched and challenging structures.
Acknowledgements
I would like to thank Dirk Schmaljohann for his help in editing this manuscript. Furthermore, I am deeply grateful to all co-workers and the agencies and companies supporting this work.