

1 Introduction
The recent period has evidenced an ever-increasing interest for chemical species of nanometric size [1]. This size corresponds to proteins, to organic macromolecules such as polymers or dendrimers or to relatively small molecules which self-organize into different sorts of objects such as micelles or vesicles. Inorganic molecular clusters [2] may also reach this size and display interesting physical properties [3]. Compared to this new approach of nanometric compounds, the inorganic particles in solution (colloids) may appear as outdated species. In fact, coloured solutions of noble metal, essentially gold, have been known for ages and the first rational synthesis of gold colloids has been described by Faraday in 1857 [4]. Many methods to produce in solution particles of either pure metal or metal oxides, and more generally chalcogenides have been reported in the literature since then [1]. These species have recently attracted a renewed interest, mainly because of their real or expected physical properties resulting from their ‘quantum size’. This concerns the fields of optics, magnetism and electronics [5]. Nevertheless, the recent renewed interest in the properties of these objects has evidenced the need for the control of the particles monodispersity, of their size, their shape, their organization and the nature of the chemical species present at their surface. In this respect, the well-known reduction methods [6] display limitations due to their lack of variability. The use of reverse micelles as ‘nanoreactors’ inside which salt reduction and particle growth occurs has allowed to obtain monodisperse nano-objects which may display a define shape (spheres, rods, wires...) and which may self-assemble on various substrates [7]. In these processes, salt and water are always in contact with the surface of the particles, thus passivating them, modifying their reactivity and, in some cases, leading to the production of oxides or hydroxides. The use of an organometallic precursor, able to decompose in mild conditions either spontaneously or in the presence of a reducing gas has appeared as a valuable alternative for the synthesis of nano-sized objects. Organometallic compounds have been used as material precursors in high temperature decomposition processes, for example in chemical vapour deposition [8]. They have also been deposited onto inorganic support to produce heterogeneous catalysts [9]. However, the high reactivity of organometallic complexes may allow a low temperature synthesis of well-defined species the chemistry and physics of which may be controlled. This approach of solution synthesis of large clusters of nanometric size has attracted recently an ever-increasing interest. Metal carbonyls have been widely used as precursors of metals either in the gas phase (OMCVD for the deposition of films or nanoparticles) or in solution for the synthesis after thermal treatment [10], UV irradiation or sonolysis [11, 12] of fine powders or metal nanoparticles. In order to be able to obtain a better control of the size and size distribution of the particles as well as to obtain clean surfaces for undertaking a reproducible chemistry, we have developed an approach based on the use of organometallic precursors able to decompose in mild conditions, generally using a reducing gas (Scheme 1 ) [13–35]. The ideal precursor is an organometallic complex containing ligands, preferentially olefinic or polyolefinic, able either to be hydrogenated to give a bare metal atom which would condense in the reaction medium or to be substituted by CO to give an unstable intermediate. The second approach, using CO, had a few precedents [36] at the time we started this research, whereas the first one, using a hydrogenation reaction, had none.
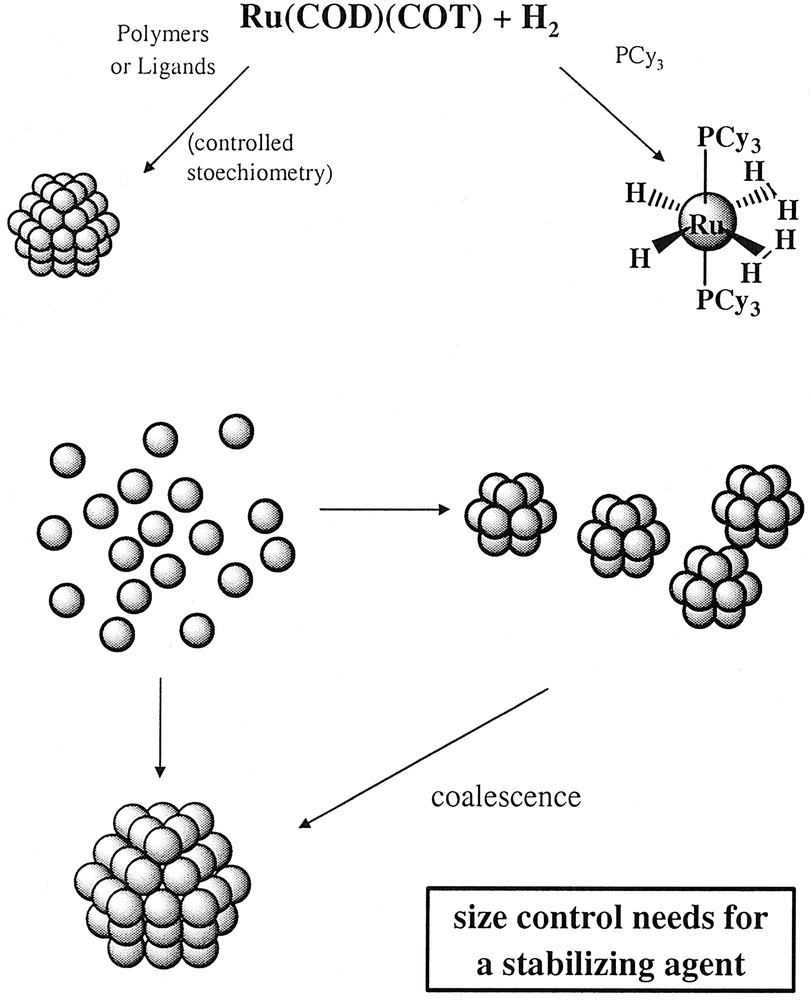
Illustration of the general synthetic method followed in our group for the synthesis of metal nanoparticles.
We will successively review hereafter the decomposition ability of the precursors, the characterization techniques employed and the synthesis of metal particles protected by polymers, ligands and organized media. We will focus this paper on the chemistry of nanoparticles of noble, catalytically active transition metals carried out in our group, essentially ruthenium and platinum, and on the development of an organometallic chemistry in solution at the surface of the particles.
2 Precursors
Purely olefinic complexes are the most attractive precursors since upon hydrogenation, they give rise to alkanes, which are innocent towards the surface of the particles. Precursors of this type are for example Ni(C8H12)2 [17] and Ru(C8H10)(C8H12) [26]. Both complexes decompose satisfactorily under dihydrogen in mild conditions. Complexes accommodating allylic groups may also decompose easily, for example Co(C8H13)(C8H12) [14] or Rh(C3H5)3. Other types of complexes may however be used when such olefinic precursors are not available. For example, M(dba)2 (dba = dibenzylidene acetone; M = Pd; Pt) [13] is a good precursor for the preparation of nanoparticles of Pd or Pt after treatment with dihydrogen or carbon monoxide. Mixed complexes such as Rh(acac)(C8H12) [32] (acac = (CH3CO)2CH) or CpCutBuNC (Cp = C5H5) [37] also decompose in mild conditions but release potential ligands of the particle namely dba, isonitrile, acacH or the corresponding diol after hydrogenation which may or may not perturb the surface.
3 Characterization methods
Whereas the methods of preparation of nanoparticles in solution using organometallic precursors are the same as those typically used in organometallic synthesis, the techniques of characterization of the products are significantly different. Thus, although it is possible in many cases to isolate and redissolve the nanoparticles like molecular species, it is not possible to obtain a crystal structure and to use routine NMR techniques to obtain a precise characterization. It is therefore necessary to combine a variety of techniques that derive from solid-state physics for the metal core of the particles and from molecular chemistry for the surface ligands.
For the characterization of the metal core of the particles, we have found in Toulouse that a satisfactory result could be obtained by a combination of two techniques, one giving precise information on one or a few objects: electron microscopy, the other being statistical over the whole sample: X-ray techniques and in particular wide angle X-ray scattering (WAXS) [38]. Transmission electron microscopy (TEM) gives the size, size distribution and shape of the particles. It can be associated to high-resolution electron microscopy (HREM), which gives the crystal structure and lattice parameters and EDX, which gives the composition. The WAXS technique provides a distribution of metal-metal bonds inside a homogeneous assembly of nanoparticles. Using a model, it is then possible to have access to the structure of the particle and to its coherence length, assuming that all particles adopt the same size and structure. In addition, powder X-ray diffraction (XRD) can allow the identification of the crystal phase for particles larger than 3 nm. More sophisticated techniques such as EXAFS may also be employed.
The second approach to nanoparticle characterization takes profit of spectroscopic techniques commonly used in molecular chemistry namely infrared, UV and NMR spectroscopy. UV has been commonly used to characterize the plasmon bands of coinage metal particles [39]. Concerning infrared spectroscopy, Bradley et al. [40, 41] have shown that metal colloids could adsorb probe molecules such as CO and that, in general, the frequency of the CO stretch was directly comparable to results obtained on monocrystals in ultra high vacuum. This may give an indication of both the particle size and the oxidation state of the particle surface. It was for example evidenced that the ratio between terminal and bridging CO groups was directly correlated to the size of palladium particles. Bradley also carried out 13C NMR studies on nanoparticles covered with 13C enriched carbon monoxide [41]. In the case of ruthenium, a metal giving rise to a very small Knight shift, and for very small particles, the presence of terminal and bridging CO could be ascertained [41]. In the case of platinum and palladium colloids, indirect evidence for CO coordination were obtained by spin saturation transfer experiments [41]. The NMR of ligand-protected nanoparticles is little developed, however, several recent studies demonstrate that it is possible to observe long chain ligands ligated to metal particles by 1H and 13C NMR spectroscopy [15, 26, 42].In this case, the nuclei close to the metal surface are not visible because of the slow tumbling of the metal particle in solution. This point will be further discussed hereafter.
4 Synthesis of metal nanoparticles embedded in polymers
4.1 Synthesis and structure
Ru(C8H10)(C8H12) reacts rapidly with dihydrogen (1–3 atm) at room temperature in a hydrocarbon solvent to give ruthenium particles and cyclooctane [26]. In the presence of a polymer (polyvinylpyrrolidone, PVP; cellulose acetate, CA), the reaction produces particles, the size of which depends upon the nature of the polymer and upon the relative concentration of the precursor and the polymer. In this way, monodisperse particles of 1 to 1.7 nm mean size have been prepared in PVP or CA according to the reaction conditions [43]. In PVP the particles display a very low size dispersity (Fig. 1 ). In contrast, the reaction in CA produces larger particles displaying a broader size dispersity, which evidences the influence of the nature of the polymer and of its coordination ability on the stabilization of the particles. The particles in PVP were analysed by HREM (Fig. 2 ) [44]. The particles adopt the hcp structure of bulk ruthenium. An interesting effect of the size reduction on the crystal lattice has been evidenced by WAXS. A contraction of 1.1% in the metal-metal distance (0.266 nm, 0.27058 in the bulk) is observed together with a relaxation of the c parameter (0.436 nm, 0.42811 in the bulk), which leads to a c/a ratio of 1.633, close to the ideal one, instead of 1.582 in bulk ruthenium.
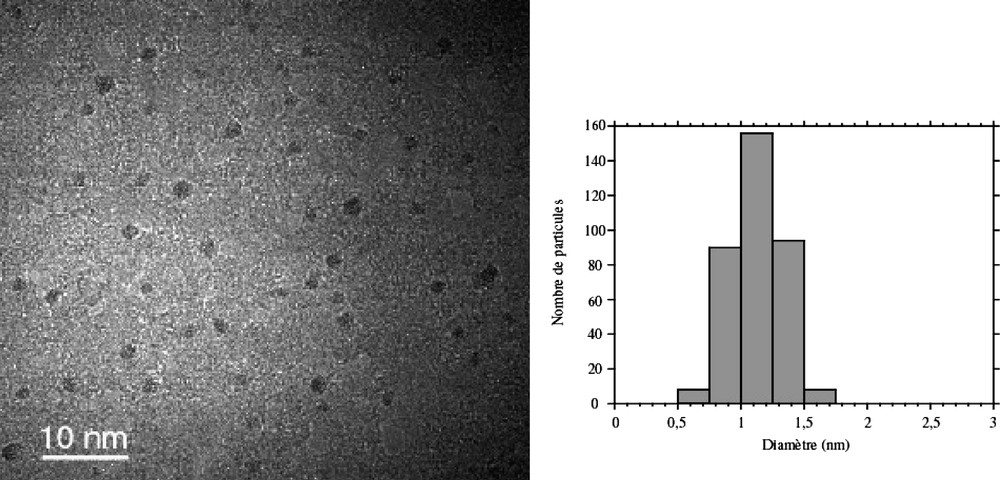
TEM micrograph and size histogram of ruthenium nanoparticles embedded in PVP.

HREM micrograph of a ruthenium nanoparticle stabilized in PVP.
Platinum colloids can be prepared in the same conditions starting from the precursor Pt(dba)2 [15]. In this case, the particles adopt the fcc structure of platinum with, in the case of particles prepared in the presence of PVP, a contraction of the Pt–Pt distance of 1.2% (0.274 nm) compared to the bulk.
In a similar way, nanoparticles of cobalt and nickel may be prepared under dihydrogen in the presence of PVP. The nickel particles display the fcc structure of bulk nickel [25]. In contrast, cobalt particles of small size (1.6 or 2 nm) adopt a non-periodic polytetrahedral structure whereas the larger ones (4 nm) adopt the hcp structure of bulk cobalt [22].
Co-decomposition of the two precursors leads to the formation of bimetallic Rux–Pt1–x particles [18, 44]. Platinum-rich particles are fcc, whereas ruthenium-rich ones are hcp. There is a critical composition Ru3Pt for which most of the particles are twinned (Fig. 3 ). In this case, the particles are totally monodisperse and very small (1.1 nm). This composition corresponds roughly to the limit of solubility of ruthenium in the platinum lattice for bulk alloys. The particles adopt a twinned fcc structure (Fig. 4 ) with the twinning wall lying in a (111) plane located in the middle of the particle (Fig. 5 ). The homogeneity in the size and shape of the twinned particles suggests a well-defined atomic organization: namely a twinned truncated octahedron for the particles, which can also be, described as well-defined clusters (Fig. 6 ).
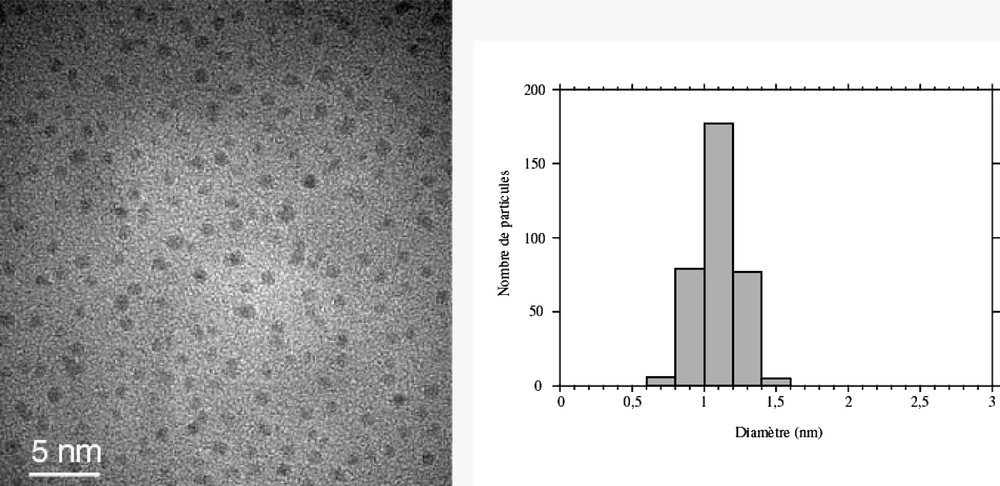
TEM micrograph and size distribution of Ru3Pt nanoparticles embedded in PVP (mean diameter ca. 1.2 nm).

RDF of Ru3Pt nanoparticles embedded in PVP obtained from WAXS analysis and comparison with a theoretical model.

HREM micrograph of a Ru3Pt nanoparticle in PVP showing the twinning (a) and image simulation (b).
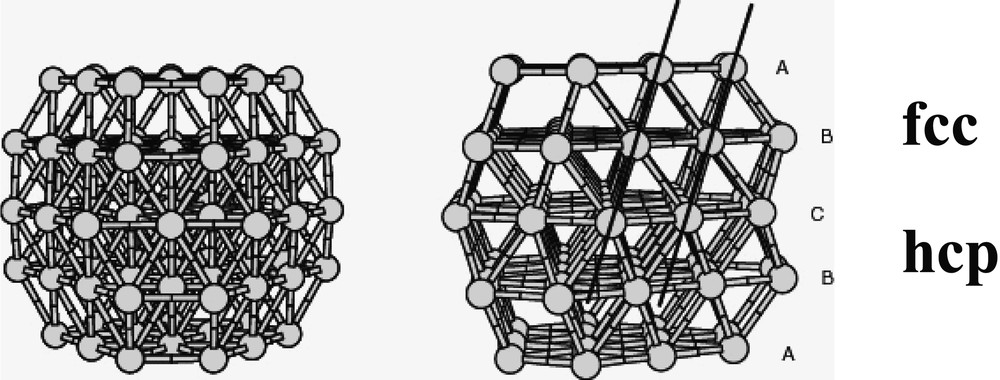
Twinned truncated octahedron model containing 79 atoms corroborating the structure Ru3Pt nanoparticles in PVP
Similarly, bimetallic cobalt–platinum nanoparticles may be prepared by hydrogenation of the two precursors Co(C8H13)(C8H12) and Pt(dba)2 in the presence of PVP [23]. In this case, according to the composition, particles of very small size adopting a non-periodic icosahedral type structure are observed. It may be interesting to note that such a non-periodic structure has recently been demonstrated by X-ray crystallography on a monocrystal for the cluster [Pd145(CO)60(PEt3)30] [45].
4.2 Surface of the particles
Perhaps the most important challenge of the field is the knowledge of the surface state of the particles. As stated in the preceding chapter, CO can be used as a probe to characterize the surface of nanoparticles. Thus, it characterizes first the availability of the surface towards coordination of incoming molecules but, furthermore, the CO stretch is very sensitive to the oxidation state of the surface and the presence of other ligands. It was found in the case of the particles prepared in PVP by hydrogenation of an organometallic precursor that the frequency observed for the CO stretch matches that observed for metal monocrystals in the ultra high vacuum. Moreover, in the case of cobalt particles, it was found that, at a given size, the particles exhibit the same increase in magnetization per cobalt atoms than those prepared by time-of-flight experiments [14]. Again, the magnetic properties are strongly influenced by the nature of the chemical species present at the surface. Both observations therefore strongly suggest the ‘clean character’ of the surface of particles prepared in these conditions.
5 Synthesis of metal nanoparticles in weakly coordinating solvents
The presence of a polymer may not be necessary for stabilizing the particles. We found out that Pt(dba)2 reacts with CO in THF to give a colloid containing fcc particles of 1.2 nm mean size and displaying a narrow size distribution [13]. This colloid is relatively stable and can further be used as a starting material to prepare ligand-protected platinum nanoparticles.
When using Ru(C8H10)(C8H12), the decomposition by H2 in neat THF or in non-coordinating solvents such as pentane leads to the precipitation of ill-defined ruthenium powder. However, when using alcohols as solvents, the reaction produces a colloidal solution containing ruthenium particles. This colloidal solution was found to be stable for long periods of time (over one year) when kept under argon. Exposure to air or addition of pentane under argon leads to the precipitation of the particles. In the latter case, the isolated particles burn in air, hence demonstrating the reactivity of their surface, whereas in the former one the particles are stable because of the formation of a passivation layer of RuO2 [19, 33].The particles prepared in neat methanol are very large, polycrystalline (ca. 76 nm) (Fig. 7 ), mesoporous and display a relatively large specific area (> 40 m2 g–1). In THF/methanol mixtures (Fig. 8 ), the size of the polycrystalline particles remains of the same order of magnitude as those in neat methanol up to a THF content of 25 vol.% after which the size decreases linearly with the THF content: for a THF content in the solution of 50 vol.%, the size of the particles is 47 nm, for 90 vol.% 20 nm and for 97.5 vol.%, ca. 3–6 nm [33]. A similar trend, namely size decrease, is observed upon changing MeOH for higher alcohols. The size of the particles is ca. 5 nm when the reaction is carried out in iPrOH and ca. 2.5 nm in pentanol. Finally, it was found in the case of the reactions carried out in MeOH/THF mixtures that addition of cyclooctane leads to an increase in the size of the particles.
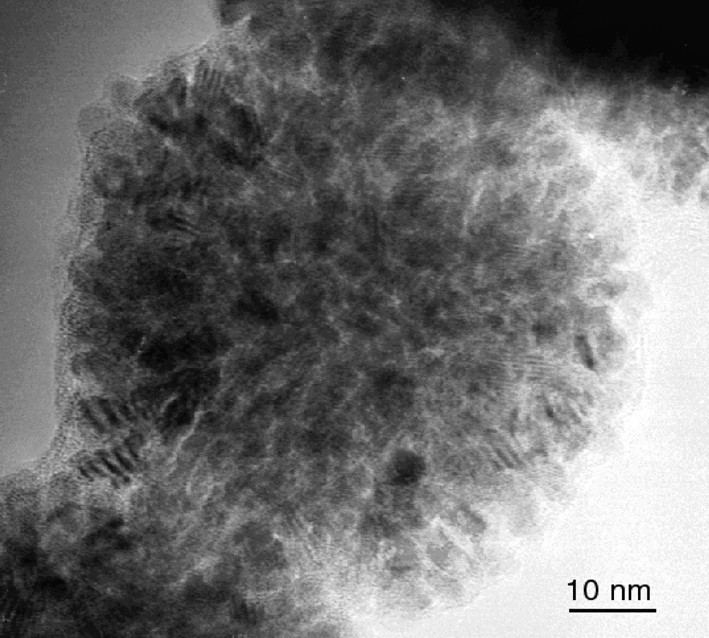
HREM micrograph of ruthenium sponge-like particles obtained in pure methanol.
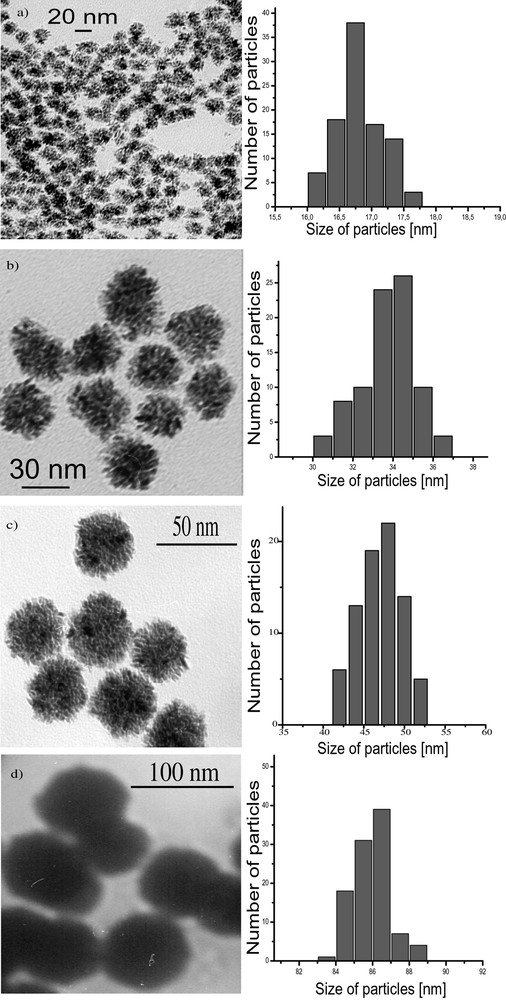
Micrographs and histograms of the size of Ru nanoparticles synthesized in different MeOH/THF mixtures (a: MeOH/THF = 5/95; b: MeOH/THF = 25/75; c: MeOH/THF = 50/50; d: MeOH/THF = 90/10).
These surprising results were attributed to the segregation in the solution between cyclooctane resulting from hydrogenation of the ruthenium precursor and the rest of the solvent (Scheme 2 ). In this respect, the larger droplets would be formed in the most polar solvent systems and hence the most segregated medium. This is in excellent agreement with the sizes of the particles measured in neat alcohols. The most lipophillic one (pentanol) gives rise to the smallest particles.
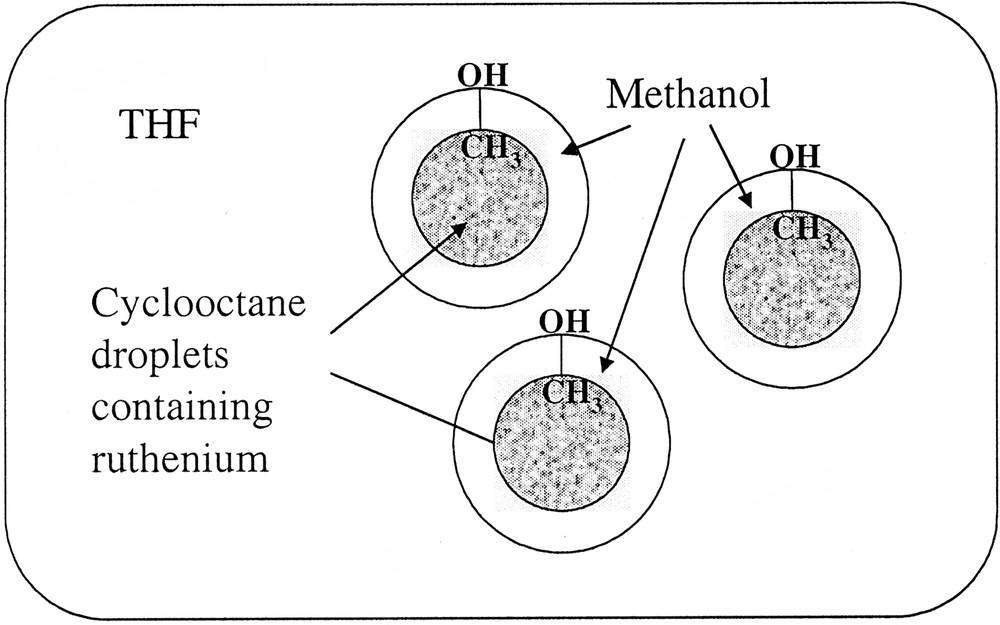
Hypothetical segregation in the solution allowing the size control of ruthenium nanoparticles depending upon the reaction medium composition.
In the MeOH/THF mixtures, the change in polarity of the medium resulting from the composition changes may account for the apparent correlation observed between the size of the particles and the MeOH content (Fig. 9 ). The mesoporous, polycrystalline nature of the large particles is surprising. It suggests that, during the growth process, nanocrystallites synthesized at the early stage of the reaction may be connected by ruthenium atoms or particles resulting from the decomposition of the remaining starting material.

Linear correlation between the MeOH/THF volume rate and the particles size.
When the reaction is carried out in heptanol [35], the particles are monodisperse in size (3 nm), well dispersed in the solvent and adopt the hcp structure of bulk ruthenium (Fig. 10 ). They can be isolated and re-dissolved in various solvents, including d8-THF for NMR analysis (Fig. 11 ). In this case, it is clear that coordinated heptanol is present at the surface of the particles and acts as a weakly coordinating ligand. In this case, the presence of surface hydrides was demonstrated by NMR techniques.
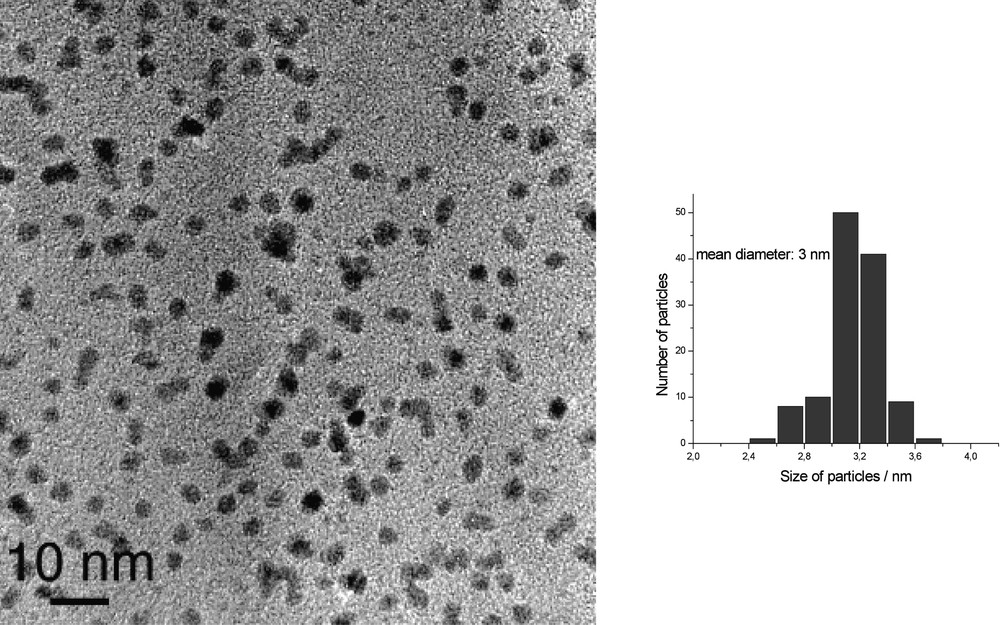
HREM micrograph and size histogram of Ru particles synthesized in pure heptanol.
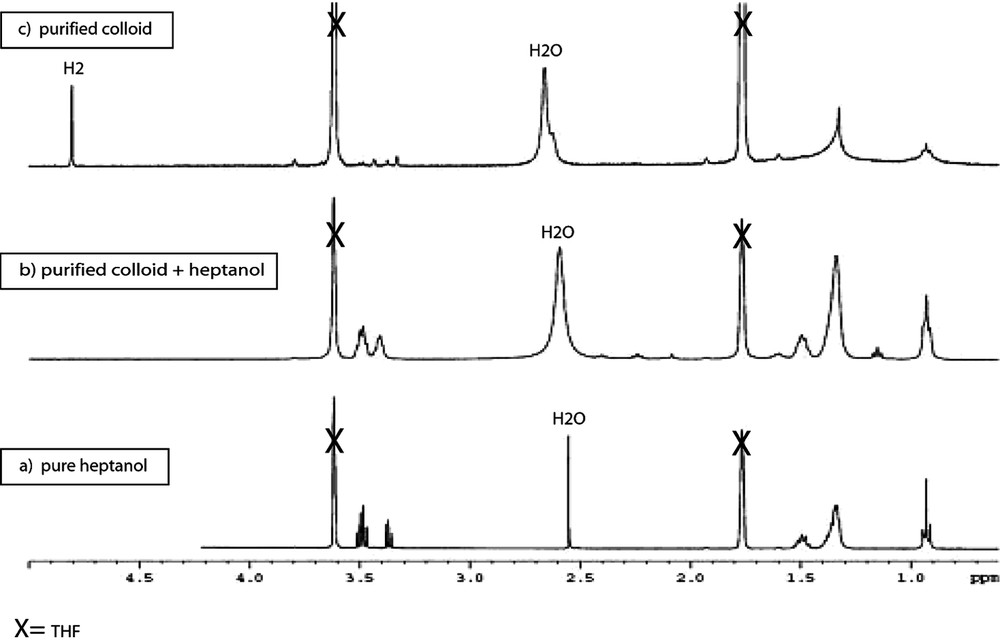
1H NMR (d8-THF; 400 MHz): (a) heptanol as reference, (b) purified Ru/heptanol colloid with an additional amount of free heptanol, and (c) purified Ru/heptanol colloid; note the presence of a sharp peak at 4.8 ppm attributed to H2 in solution.
6 Synthesis of ligand-protected metal nanoparticles
Chemical species able to stabilize the surface of metal particles in solution have been widely used for a long time. Classical methods of colloid preparation use this approach; for example, the Turkevich method [46] of synthesis of gold colloids uses citrate ions as surface stabilizers. Giant clusters, the most famous one being Au55Cl6(PPh3)12 prepared by G. Schmid [47], also take profit of organic or inorganic ligands to stabilize a species containing a large number of metal atoms.
6.1 Monodentate ligands
The methods described above to prepare metal nanoparticles embedded in polymers can be transposed for synthesizing ligand-protected particles. For example, Pt(dba)2 reacts with CO (1 atm) in THF to give fcc particles of 1.2 nm mean size. The same reaction carried out in the presence of 0.2 equiv PPh3 leads to phosphine protected particles which may also be prepared by addition of PPh3 to the preformed colloid and which adopt an icosahedral structure [13]. Interconversion between the two colloids is possible by addition of CO on one side or of PPh3 on the other. These experimental procedures are not specific of the phosphine ligands and may be employed using octanethiol [15]. In this case, a very stable colloid is formed. It may be stored under argon for a long time without alteration and re-dissolved for reactivity or spectroscopic studies. The colloid contains monodisperse fcc particles of 1.6 nm mean size (Fig. 12 ). WAXS studies demonstrate the strong fixation of the ligand at the surface of the particles (Fig. 13 ). This is corroborated by IR (Fig. 14 ) and solution NMR studies which point to the absence of exchange between free and coordinated ligands at the NMR time scale. In this case it is possible to observe by 13C NMR the carbon atoms of the alkyl chain of the coordinated ligand as broad peaks except for the carbons located in α, β and γ position relative to sulphur, which are too broad to be observed.

TEM (a) and HREM (b) micrographs of octanethiol stabilized platinum colloid showing nearby particles with no coalescence.
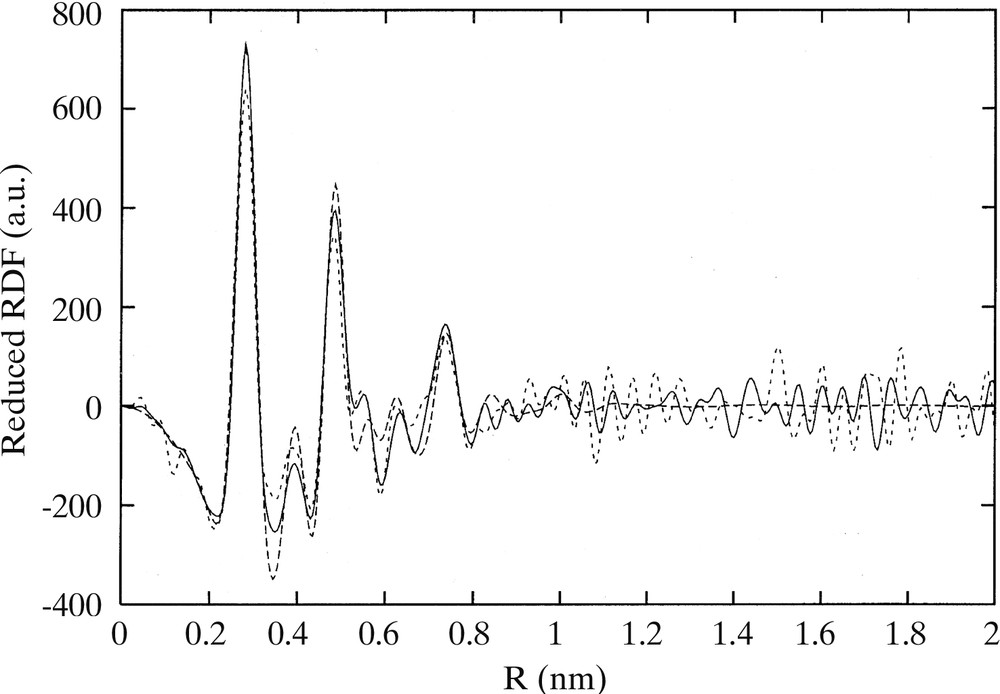
RDF of octanethiol stabilized platinum colloid obtained from WAXS analysis.

IR spectra of Ptx(CO)y(THF)z and Ptx(CO)y(THF)z(C8H17-SH)w colloids.
It is possible to take profit of the strong coordination of sulphur to platinum to build ‘supramolecular’ networks incorporating the metal particles. Thus platinum particles of 1.6 nm mean size can be prepared using as protecting ligands thiophenols substituted in the 4 position by a hydroxo, a carboxylate or an amino group [29]. Self-assembly of the nanoparticles is observed for the colloids stabilized by 4-HO–C6H4–SH or 4-HOOC–C6H4–SH. If a mixture containing a 1:1 mixture of the 4-HO–C6H4–SH and 4–H2N–C6H4–SH is used, the particles self-organize into very long nanotubes the walls of which are constituted by a monolayer of platinum nanoparticles (Fig. 15 ).

TEM and HREM micrographs of platinum super-structures resulting from the assembly of nanoparticles stabilized by a 1:1 mixture of the 4-HO–C6H4–SH and 4-H2N–C6H4–SH at different magnifications: (a) × 15 000; (b) × 100 000; (c) × 600 000.
It is noteworthy that when using hexadecylamine (HDA) instead of a thiol, particles in the same size range (1.6 nm) initially form but coalesce into worm-like structures and nanowires (Fig. 16 ) [48].

TEM micrograph of hexadecylamine stabilized platinum nanoparticles, showing the formation of nanowires.
The reaction of Ru(C8H10)(C8H12) with dihydrogen (1–3 atm) can also be carried out in the presence of long chain thiols or amines [26]. In both cases hcp particles displaying a low size dispersity and a mean size of 2–3 nm are obtained. The particles prepared in the presence of thiols appear agglomerated by TEM studies, whereas the one prepared in the presence of amines are more regularly dispersed on the microscopy grid. In both cases, it has been possible to isolate the particles and dissolve them again for NMR studies.
The 1H and 13 C NMR spectra of octane thiol protected ruthenium particles are very similar to those obtained for the platinum/thiol colloids described previously (Fig. 17 ). However, upon addition of an excess of thiol, it is possible to observe the catalytic transformation of thiols into disulfides. This is not a simple oxidation but rather a coupling reaction since traces of dioxygen block the reaction and since dihydrogen has been detected in the NMR tube.
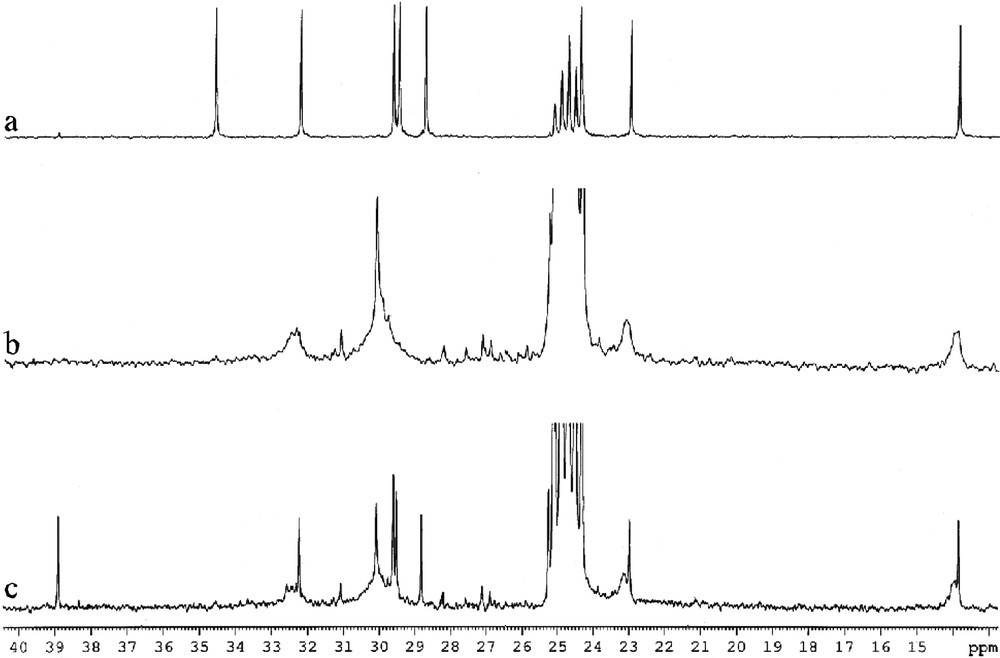
13C NMR spectra (d8-THF, 101 MHz) of C8H17SH (a), C8H17SH-stabilized Ru colloid (b) and C8H17SH-stabilized Ru colloid + excess C8H17SH (c).
This result demonstrates the availability of the ruthenium surface for a catalytic reaction implying a succession of oxidative additions and reductive eliminations.
In the case of the particles accommodating amine ligands, a new phenomenon has been evidenced, namely a dynamic exchange at the NMR time scale between free and coordinated amines (Figs. 18 and 19 ). It has been correlated to the TEM and HREM results which show that, at the early stage of the reaction, the particles display a spherical aspect and a small size (ca. 2–3 nm) and that after a few hours the particles coalesce into elongated wormlike particles still constituted of pure, unoxidized hcp ruthenium (Figs. 20 and 21 ). The NMR observation is particularly interesting since it shows that these particles behave like molecular clusters for which such phenomena are frequent.
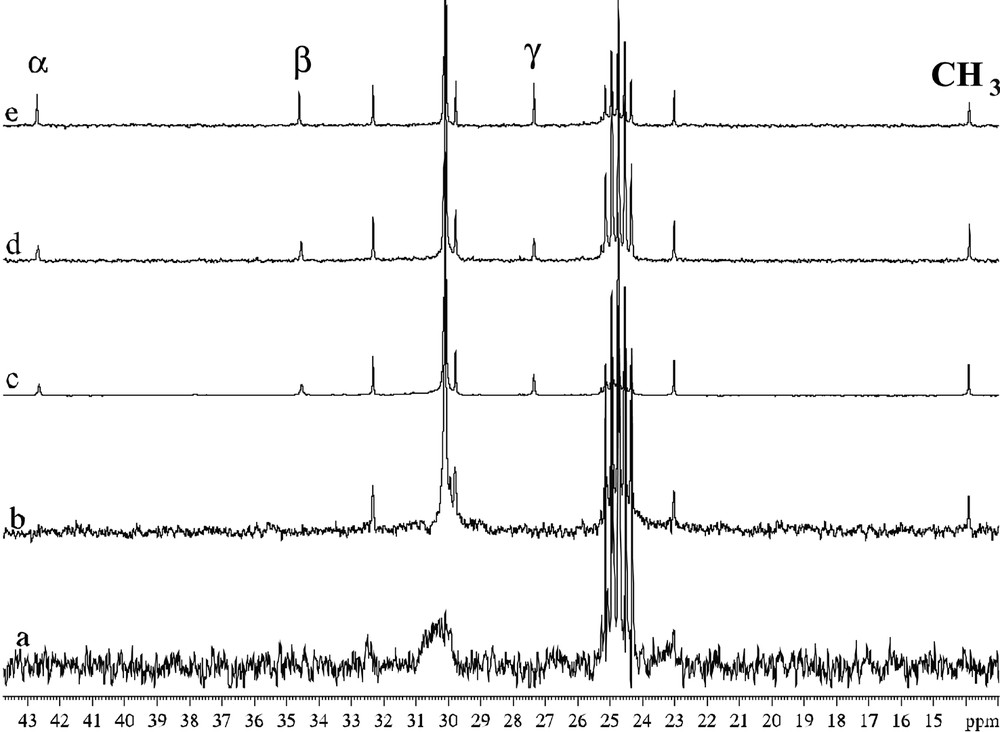
13C NMR spectra (d8-THF, 101 MHz) of C16H33NH2-stabilized ruthenium colloid (a), C16H33NH2-stabilized ruthenium colloid + excess C16H33NH2 (b,c,d), C16H33NH2 (e).

Enlargement of Figs. 16b–e for comparison of linewidths.

TEM micrograph of C16H33NH2 stabilized ruthenium colloid (0.2 equiv) at low concentration in THF, showing their spherical morphology.
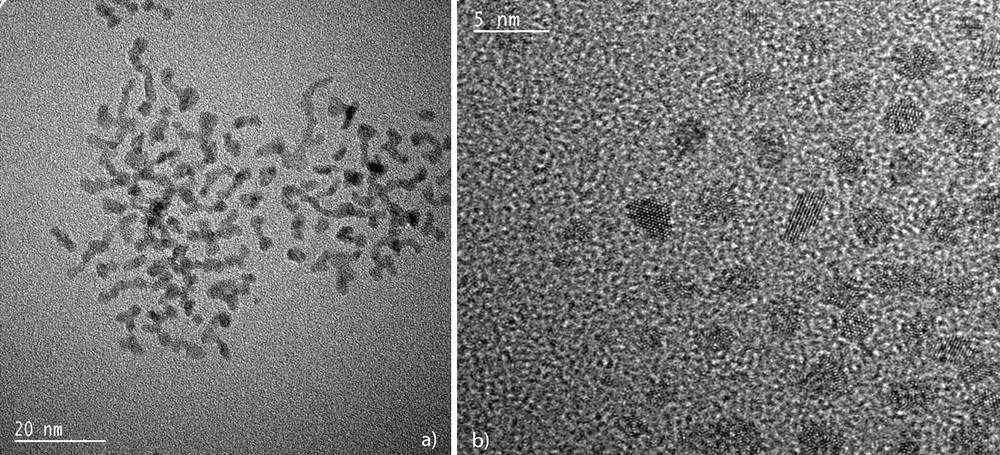
TEM (a) and HREM (b) micrographs of hexadecylamine-stabilized ruthenium nanoparticles (mean size ca. 2–3 nm) showing the elongated aspect of the particles.
6.2 Bidentate ligands
The synthetic method described above is very simple and can be in principle transposed to any metal/ligand combination. One of the most challenging problems regarding the use of nanoparticles in catalysis concerns asymmetric catalysis. Only a few examples of asymmetric heterogeneous catalysis have been reported, the most popular ones using a platinum cinchonidine system [49].
Nanoparticles of both ruthenium and platinum can be prepared using asymmetric oxazolines or amino alcohols as ligands (Scheme 3 ) [34]. In both cases, the ligands provide an excellent stabilization of the particles that can be handled like molecular species. The platinum particles give rise to self-organized super-structures adopting shapes of wires (Fig. 22 ) or of pseudo-crystals (Fig. 23 ). Scheme 4 illustrates the interactions that may exist between ligand molecules (hydrogen bonds) at the surface of the particles undergoing this type of organization. The ruthenium particles are very small (1 to 2 nm according to the ligand) and can be used in catalytic reactions such as asymmetric hydrogenation of dimethylitaconate (40 bar H2, MeOH, rt) or asymmetric hydrogen transfer from isopropanol to acetophenone (tBuOK, rt). In this case a distinct reactivity has been found using the asymmetric oxazoline shown in Scheme 3 between molecular species and nanoparticles. The nanoparticles are much more active but much less selective than the corresponding molecular complexes. Thus, a very modest enantiomeric excess (10%) has been found which suggests that asymmetric catalysis can indeed take place on such large chemical species [50].
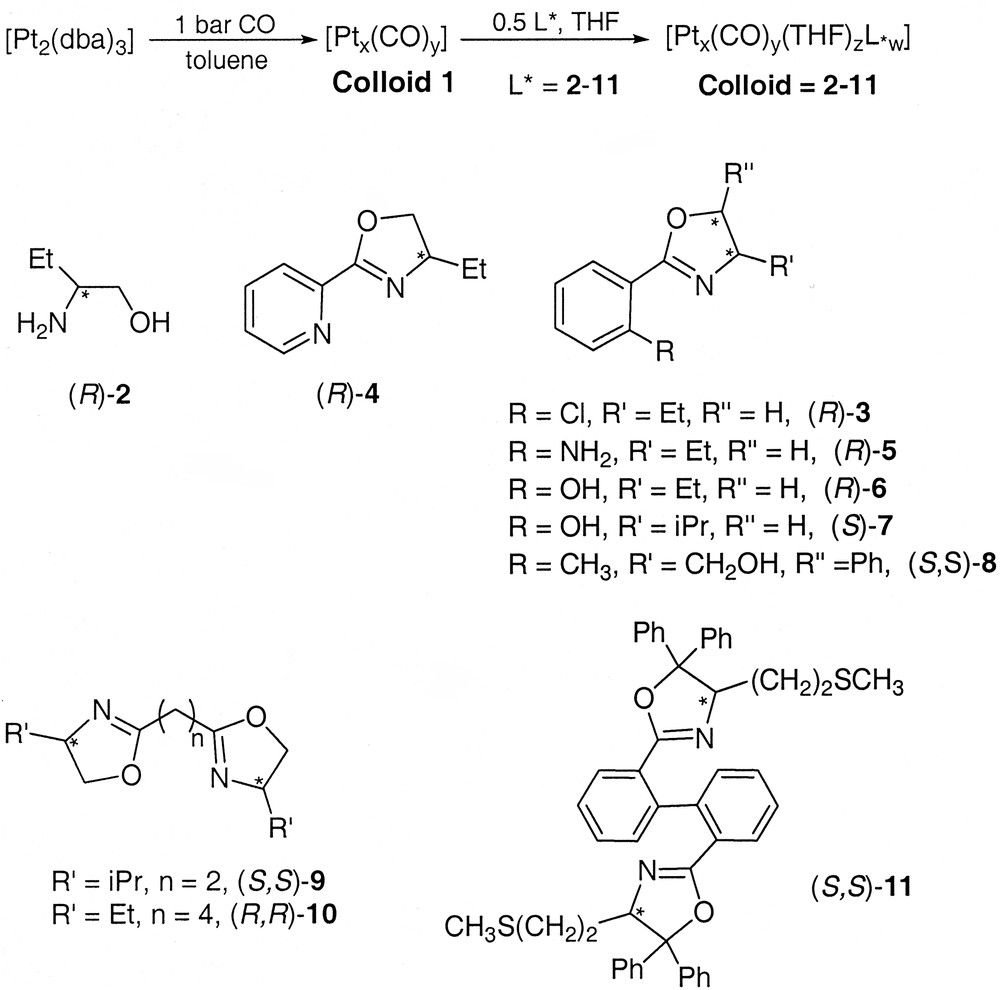
Colloids synthesis with ligand structures.

TEM micrographs of oxazoline stabilised platinum particles showing the platinum nanowires and their assemblies in bundles (the inset of (a) is presented at a higher magnification in (b)).

TEM micrograph of oxazoline stabilised platinum particles showing nanocrystals (a) and an enlargement of a 2-nanocrysatl junction (b).

Illustration of possible interactions between particles through strong hydrogen bonds between ligands.
Finally, we found recently that diphosphite stabilized palladium nanoparticles were active catalysts for asymmetric allylic alkylation reactions. More exactly, the reaction of rac-3-acetoxy-1,3-diphenyl-1-propene (rac-I) with dimethyl malonate under basic conditions was studied using a Pd colloid stabilized by a chiral xylofuranoside diphosphite. The palladium particles have been shown to be less active than the molecular counterpart, but excellent enantioselectivity (> 99%) was obtained. In addition, a total selectivity for the alkylation of only one of the enantiomers of the substrate has been observed. These results reveal that metal nanoparticles can behave differently from the reactivity currently found in molecular catalytic systems [51].
7 Conclusion
In summary, we have described above our approach towards the synthesis of novel nano-objects consisting of a metal core and a surface which may be functionalized by addition of organic ligands. TEM pictures of the metal core of these nanoparticles appear similar to those of particles commonly used in heterogeneous catalysis or to colloids prepared by well-known reduction methods. However, the organometallic approach displays several specificities, which can be summarized as follows.
- • The organometallic approach is a low temperature approach, which means that in general the synthesis of the particles and therefore many characteristics of the nano-objects can be controlled:
- ◦ the size and size dispersity of the particles,
- ◦ the structure of the particles, which may be different from that thermodynamically stable in the reaction conditions : for example, the polytetrahedral structure found for cobalt nanoparticles,
- ◦ the composition in case of bimetallic species.
- • The organometallic approach allows the control of the surface. It is therefore possible to prepare species displaying a clean surface able to adsorb small molecules (CO, H2) or large ligands (phosphines, amines, thiols, polydentate ligands, etc.).
- • The surface properties of these nano-objects match those of metal nanocrystals prepared in ultra-high vacuum, for example the C–O stretch of adsorbed carbon monoxide or the magnetic properties of cobalt particles embedded in PVP. This demonstrates the ‘clean’ character of the surface of these particles and their availability for reactivity studies.
- • These nano-objects display an organometallic surface chemistry comparable to usual organometallic moities and which can be studied by classical spectroscopic methods, for example : substitution reactions leading to structural changes in the particles, the fluxional or non fluxional behaviour of surface ligands, the formation and observation of surface hydride species, the monitoring of catalytic reactions etc.
- • These species display a very rich potential of reactivity, which may concern fields as diverse as dihydrogen formation and storage or asymmetric catalysis.
- • Finally, the shape and self-assembly of these particles can also be controlled, which gives rise to novel nanomaterials displaying interesting physical properties in the fields of semi-conductors, magnetism or optics [24, 25, 27, 31].
All these elements suggest therefore that there is a strong potential for organometallic chemists to enter this research area, which concerns the synthesis and properties of metal nanoparticles. This should lead to impressive developments in the field of surface organometallic chemistry in the future.
Acknowledgements
The authors thank the students and colleagues who have participated to the work over the years. In addition, we gratefully acknowledge CNRS, MENRT, EC, and TMR network CLUPOS for support.