

1 Introduction
Colloid and surface science and technology often make use of monolayers of organic molecules that adsorb on particles and surfaces of metals (free metals, oxides and chalcogenides). These monolayers are applied to modify the surface properties, to functionalize the particles and surfaces, to stabilize them with regard to unwanted chemical reactions and aggregation processes of colloids, to imprint onto them an ‘address’ and to assemble them. Alkylthiols and disulfides first studied for their self-assembly properties on metal surfaces [1–8] and later were shown to be highly effective and rather global stabilizers and modifiers of metal-containing colloids [9–21]. In a similar fashion, oleate and other fatty acids were used [22–23].
The present account discusses yet another and little explored family of useful compounds, alkyl xanthates – and related materials such as thiocarbamates and trithiocarbonates – (Fig. 1), as convenient and highly versatile reagents in colloid science of metal, metal chalcogenide and metal oxide colloid science and technology. We demonstrate here that xanthates can serve in two major and central roles: They can serve as efficient capping agents for stabilizing metal-containing nanoparticles and modifying their surface properties. But, uniquely, they also can be utilized as convenient precursors and as a source of sulfur for the production of metal sulfide nanoparticles of high quality.
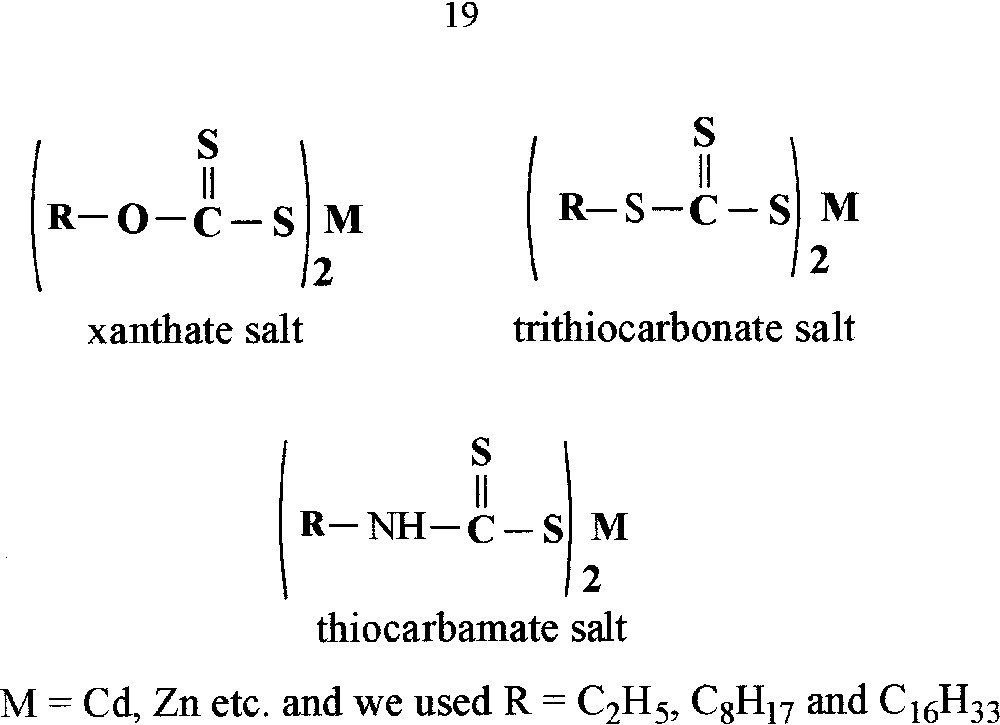
The metal salts of xanthate, trithiocarbonate and thiocarbamate.
Xanthates are proposed as useful general tools in the growing arsenal of nanotechnology and colloid and interface science and technology. They complement thiols, carboxylates and amines [1–23]. As we shall show xanthates possess unique properties that provide new possibilities. These special properties derive mostly from the relative instability of xanthates compared to thiols, carboxylates and amines. This imparts to xanthates (and to the related compounds) a measure of reactivity, which is at the core of their application as precursors and as controlled temporary capping and protecting agents. Furthermore, unlike thiols, they are charged and thus show a much-improved solubility in water or polar solvents.
The chemistry of xanthates is dominated by the –X–CS2 functionality (X = O, NH or S for xanthates, thiocarbamates and trithiocarbonates, respectively) [24–26]. Xanthates are produced cheaply in large volumes. In the laboratory alkylxanthates are produced by reacting the corresponding alcohols with CS2 [27]. Xanthates can be kept at ambient conditions. At readily attainable temperatures (~80–100 °C), xanthates can revert back to the alcohols in the presence of water.
The most important reaction from our standpoint is the well-known Chugaev reaction [24]. A related reaction might be involved in the synthesis of sulfides from metal xanthates. In a series of reports Woods et al. [28–31] discussed the redox interaction of adsorbed xanthates with coinage metal electrodes utilizing Surface Enhanced Raman Spectrocospy [32]. Zard et al. [33] investigated reductive cleavage of xanthates. Many studies investigated the uptake and extraction of metal ions by xanthates (some recent examples are in [34–35]). Xanthates (such as ethyl xanthates) are widely used as collectors in flotation in the mining industry [28–31].
In pioneering recent work, O’Brien et al. demonstrated the use of several xanthates in the synthesis of CdS nanoparticles [36]. We will discuss this work in this account, along our own work [37].
In this brief account we describe two main aspects of xanthates useful in surface and colloid science and nanotechnology. First we discuss the capping and stabilization of silver and copper nanoparticles [38] and describe the stronger interaction of xanthates with Pt and Au colloids [39]. Next we outline very recent work that uses xanthates as precursors for single-pot synthesis of high quality fluorescing metal sulfide particles, such as CdS [37] and CdS/ZnS core-shell particles. Some of the results we report are entirely new, and some are still a part of ongoing projects. We end this report with a short discussion of some future developments.
2 Xanthates as capping agents for stabilizing silver, copper, gold and platinum colloids [27, 38]
‘Non-coated’ and ‘coated’ sols are prepared as described in detail elsewhere [22, 27, 38]. Capping by the xanthates is carried out by adding appropriate (methanol or water) solutions of the capping agent to aqueous suspensions of a pre-formed ‘non-coated’ colloid. The transfer of the xanthate-capped colloids to an organic media is obtained spontaneously, by stirring equal volumes of the aqueous capped colloid solution and the organic liquid, usually with the addition of NaH2PO4. Capping by oleate is achieved by adding sodium oleate prior to the silver reduction or subsequent to it. Thiols are applied by stirring together the respective hydrosol with a tetrahydrofurane solution of the thiol.
2.1 Xanthate-capped silver hydrosols [38]
Fig. 2 shows that adding C16 xanthate to a silver hydrosol redshifts the ~400 nm UV-visible absorption by 15–20 nm and slightly broadens it, as observed directly from the yellow to orange color change.

UV-visible spectra of native and C16xanthate-capped silver hydrosols.
A typical electron transmission micrograph (TEM) of the xanthate-coated colloid is shown in Fig. 3. Mostly spherical particles are observed with a radius of 5±1 nm.

Transmission Electron Micrograph of a C16xanthate-capped silver hydrosol.
2.2 Formation of xanthate-capped silver organosols [38]
Silver xanthate readily transfers into chloroform, dichloromethane and 1,2-dibromoethane typically with 50% efficiency. Coating of the walls of the vessel accounts for the missing silver. Fig. 4 shows the spectra measured in the aqueous phase and that of the organosol, prior and subsequent to the particle transfer to chloroform. Xanthates, unlike thiols, are not sufficiently hydrophobic to enable transfer into dodecane.

UV–visible spectra of C16xanthate-capped silver colloid transferred from water to chloroform.
2.3 Silver colloid stability [38]
Xanthate-capped silver hydrosols and organosols are stable for a few weeks. The xanthate molecule itself decomposes to a significant degree under the same conditions and might be the major reason for the instability of the colloid. In comparison, a ‘non-capped’ silver colloid or an oleate-capped hydrosol is stable over at least a few months, and their organosols are stable practically indefinitely. Silver colloids protected with a hexadecane thiol (C16SH) layer in organic phases are also more stable than the xanthate-capped particles, though a 30% decrease of the extinction is observed in one month.
‘Non-capped’ silver colloids are completely stable to heating for 3 h (at 96 °C), while the xanthate-capped hydrosols and organosols show significant precipitation within 1 h, which is practically complete in 3 hours (Fig. 5). Indeed the xanthate thermally decomposes under these conditions within 1 h. Thus, the stability of the xanthate-capped colloid is determined essentially by the time and temperature instability of the capping agent itself. Instability is undesirable when one seeks long-range stability or heat resistance, however, it can be handy for use as a disposable colloid. Likewise oleate-capped silver hydrosols also deteriorate by heating at 96 °C, though more slowly than xanthate-capped particles. In contrast, thiol (C16)-stabilized silver colloids in dodecane are quite immune to the heat treatment.
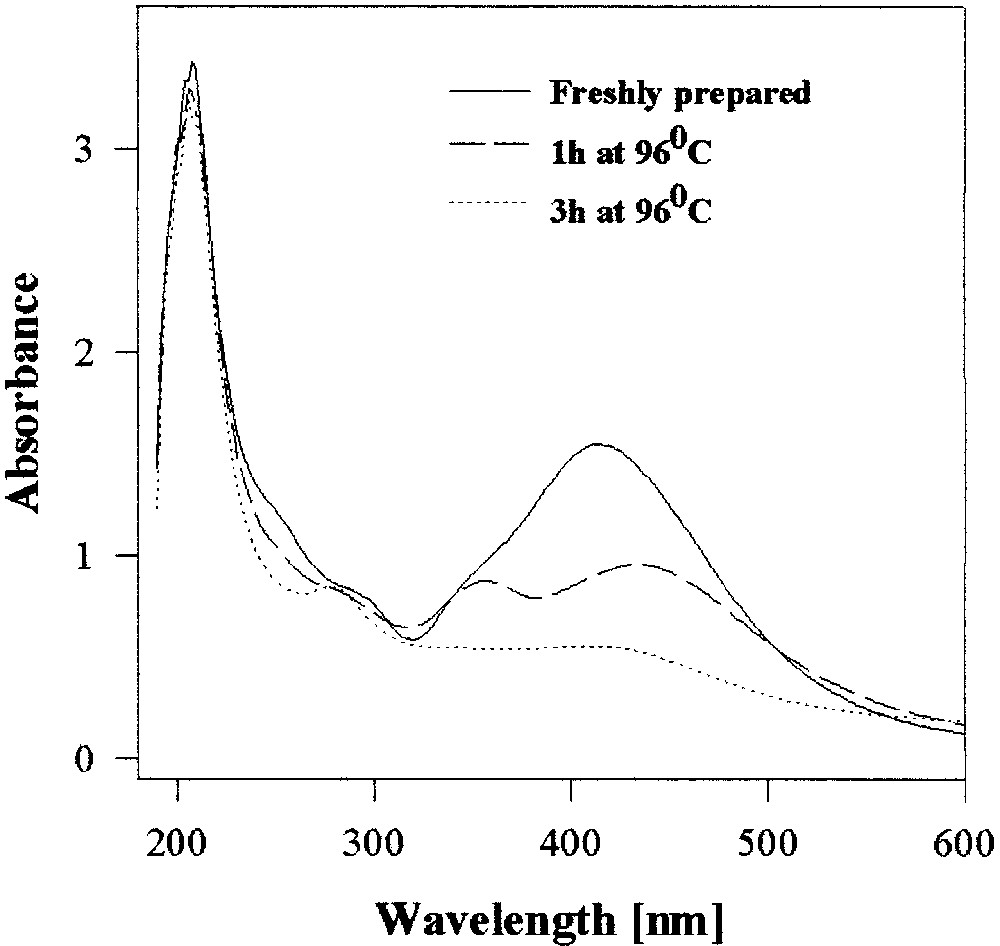
UV–visible spectra of heat-treated C16xanthate-capped silver hydrosol.
Chemical inertness of the particles is another important aspect of stability. We use the duration of oxidation by oxygen (and eventual dissolution) induced in the presence of cyanide ions as a measure of this stability.
Adding a solution of potassium cyanide to a ‘non-capped’ silver hydrosol results in a total dissolution of the colloid within 5 s. In comparison, under the same conditions a xanthate-capped silver hydrosol is highly stable. Even after 4 days most of it is retained (Fig. 6).
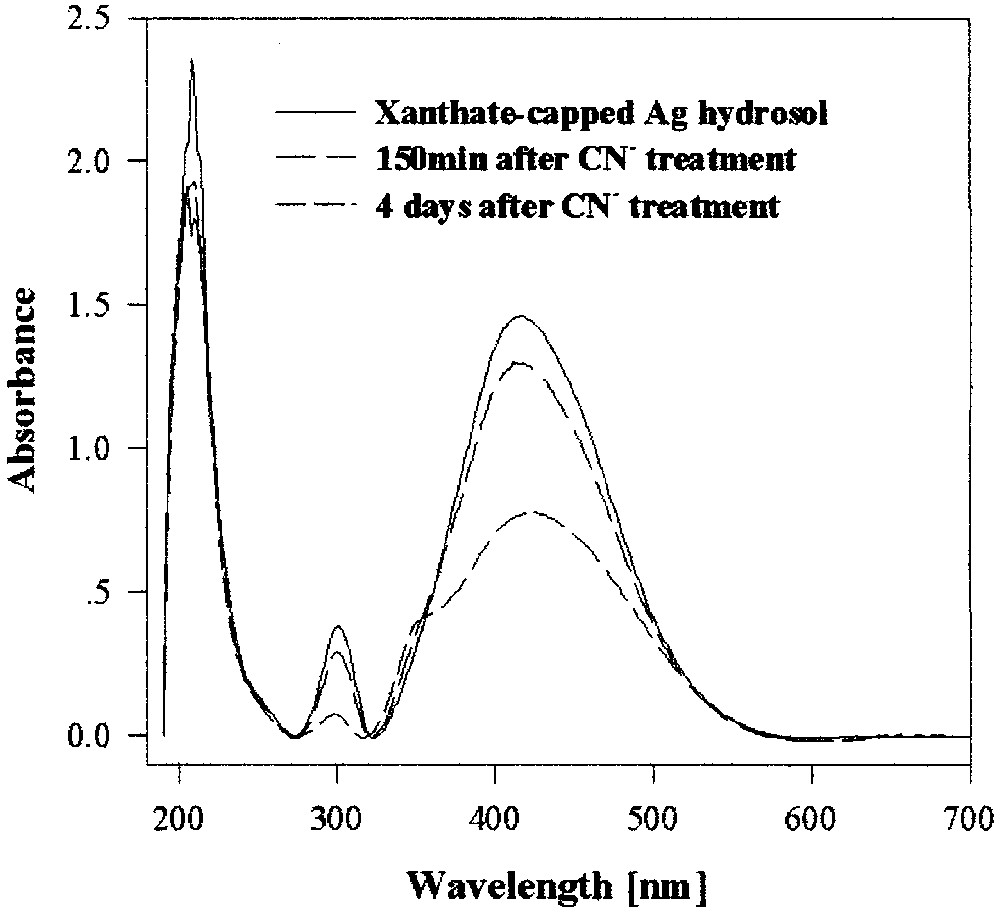
UV–visible spectra of a xanthate-capped silver hydrosol treated with cyanide in aerated solutions.
Capping by oleate, too, increases the resistance of the colloids towards oxidation in the presence of cyanide (and totally disappear after 5 h of cyanide treatment), but to a lesser degree than that exhibited by the xanthate. Alkanethiol-capped silver colloids do not disperse in water, so a comparison to them is not meaningful.
Ligand exchange is another measure of relative chemical stability of the capping layers. We follow the exchange by measuring the kinetics of cyanide-induced oxidation of the capped particles, based on the differences noted above. Starting from an oleate-capped silver organosol, we find that both xanthates and thiols readily replace the oleate, as observed by the significantly improved resistance to corrosion.
To summarize, on the one hand, alkylxanthate capping renders silver particles exceptionally stable to chemical attack (corrosion by oxygen) compared to ‘bare’ or oleate-capped particles. Xanthates also replace oleate adsorbed to the particle surface. On the other hand, xanthate capping introduces sensitivity to temperature (and time) because of the degradation of the xanthates themselves, opening new potential uses as temporary capping and disposable colloids.
2.4 Xanthate capping of copper, gold and platinum colloids [27, 38]
The general behavior of copper colloids in the presence of alkylxanthates is similar to that of silver. Yet, it is more striking since copper colloids (especially hydrosols) are notoriously unstable. Xanthate capping enables the production of copper particles stable over periods of weeks, and immune to corrosion, even in the presence of cyanide.
C16-xanthate-capped copper particles in water are stable over a fortnight, while the ‘bare’ colloid oxidizes and precipitates within minutes. The xanthate-capped copper colloid spectrum (Fig. 7) exhibits a peak at 400 nm, characteristic of small copper particles (with a radius smaller than 4 nm [39] and consistent with TEM studies [38].
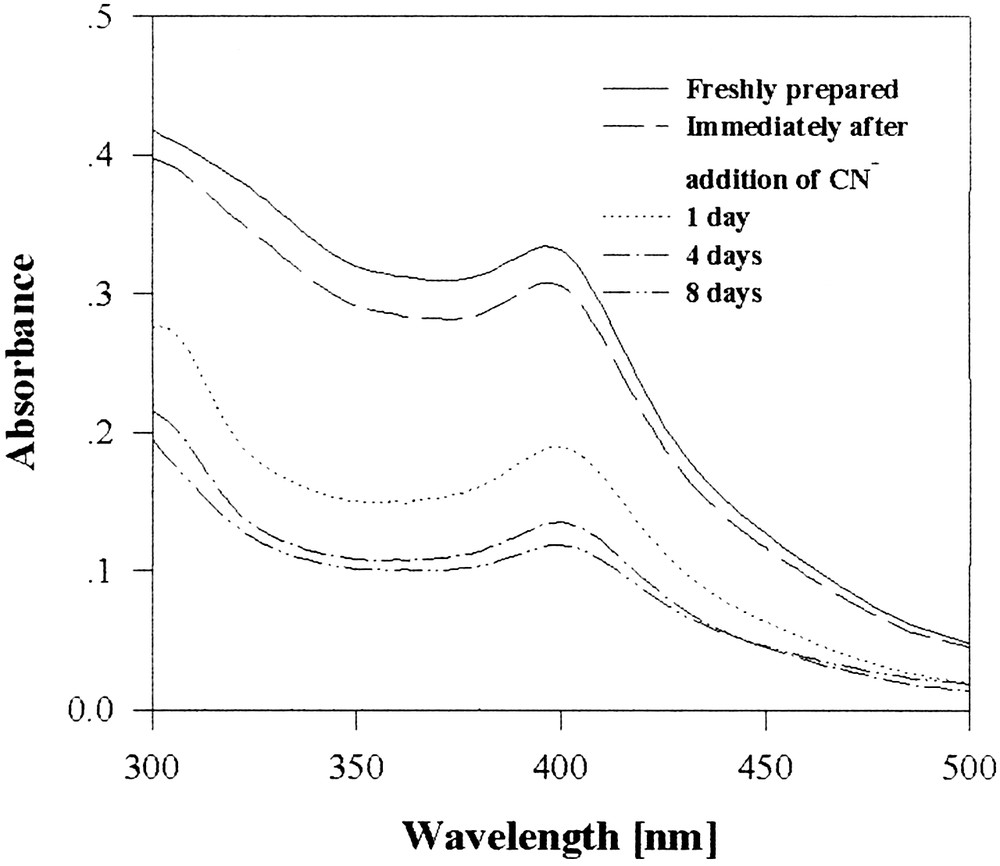
UV–visible spectra of a xanthate-capped copper hydrosol before and after treatment with cyanide.
The xanthate-capped copper colloid is by-and-large resistant to oxidation in the presence of cyanide ions, remaining mostly intact even after 8 days. In comparison, under the same conditions, a non-capped metallic copper precipitate totally oxidizes in the presence of cyanide within 2 s.
Similar to silver colloids, the xanthate-capped copper colloid transfers into chloroform or dichloromethane with 50–70% efficiency by adding sodium phosphate, and does not transfer into dodecane. Instead, it forms an interfacial colloid at the dodecane/water interface. This suggests that the xanthate-capped copper colloid (similar to xanthate-capped silver colloids) is not as hydrophobic as the thiol-capped particles.
The general behavior of xanthate-capped gold colloids agrees with that of silver and copper, demonstrating the generality of xanthate capping.
At low xanthate/Au molar ratios (~0.04) a broad feature at ~700 nm appears and grows in time (Fig. 8), the color changes from red to purple and then to blue, and the gold colloids do not transfer into organic phases anymore. This is due to partial aggregation of the colloid, as seen previously for thianicotinamide on a gold sol [40]. When higher xanthate concentrations are used (with a xanthate/Au molar ratio of ~10 and higher), this aggregation is prevented.
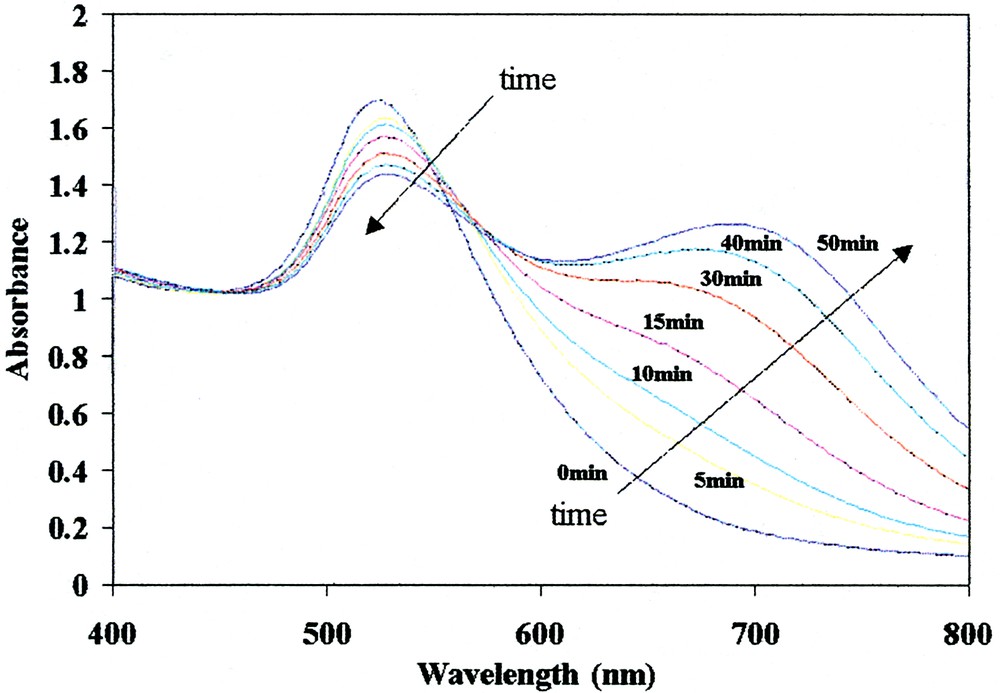
UV–visible spectra of a C8xanthate-capped gold hydrosol at Au/xanthate molar ratio of 25 as a function of time.
In almost all respects, platinum colloids behave similarly to the silver, copper and gold colloids when subjected to xanthate treatment. A unique feature of xanthate-capped platinum particles is the appearance of a strong ‘molecular’ interaction of xanthate and platinum surface atoms, as indicated in the UV–visible absorption spectra [27].
3 Xanthates as precursors for metal sulfides
3.1 General discussion of xanthate-based metal sulfides
Very recently, O’Brien et al. [36] and our group [37] showed that xanthates can be very useful in producing various metal sulfides particles, such as fluorescing CdS nanoparticles.
In pioneering work, O’Brien et. al. [36] recently reported the production of CdS particles by heating a Cd xanthate salt (using ethylxanthates) at ~200 °C, in TOP/TOPO (trioctylphosphine/trioctylphosphine oxide) as solvent following well-established methods utilizing this solution for the production of various metal chalcogenide particles [41–43]. Furthermore, the CdS particles fluoresce upon excitation in the UV via surface defect states (deep traps [44]) exhibiting a broad emission band in the red.
Independently we, too, developed a new general and versatile method to produce a variety of metal sulfides (CdS, ZnS, CuS, MnS, PbS, etc.) from their respective xanthates in a single-pot, single-precursor manner [37]. Unlike previous work, which often utilizes TOP/TOPO solutions, our method makes use of alkylamines (hexylamine, dodecylamine, octylamine, etc.) as the solvent. The strong Lewis base nature of these solvents enables a low-temperature (as low as 70 °C for CdS and even room temperature for HgS and PbS) decomposition of the metal xanthate, yielding high-quality nanoparticles. This is perhaps the first demonstration of the production of crystalline particles at such low temperatures. Our method has the following advantages.
- • The particle sizes are easily controllable in the diameter range 3–6 nm by the reaction temperature, the reagent concentration, the specific solvent, the specific chain length of the alkylxanthate size (thiocarbamates or trithiocarbonates) or the reaction time.
- • At each size, the particles form in a narrow size distribution (diameters typically varying by less than ~ ±0.4 nm)
- • This single-precursor synthesis can be carried out in the ambient, at conveniently low temperatures and without taking any mandatory precautions to exclude oxygen or humidity (a glove box is not necessary).
- • One can use the same compounds and the same procedure to produce core and core/shell (and core/shell/shell) structures.
- • The same method generally applies to many different metals and metal sulfides, with only small changes in the specific reaction conditions (which potentially enables producing core/shell structures in a single-pot manner).
- • When relevant (for CdS particles, for instance) the particles are highly fluorescing. Specifically, CdS particles exhibit an intense and very narrow (FWHM ~25–35 nm) excitonic emission, with a quantum yield of a few percent. CdS/ZnS particles produced via the xanthate method have a ~15% quantum yield at the excitonic band.
- • The precise position of the excitonic emission band (and the absorption threshold) can be tuned in steps of ~10 nm by controlling the particle size.
- • The intensity of the longer wavelength and broad emission from surface (deep) traps can be controlled. One can obtain particles whose fluorescence is dominated by the defect emission, while under more careful control and annealing this band can be removed, leaving only a strong excitonic luminescence. Dual band particles (excitonic/surface defect) can be produced.
- • All these different particles can be capped with a variety of organics without affecting their emission properties.
We demonstrate the xanthate-based method by discussion of CdS and ZnS particles and core/shell structures.
3.2 CdS particles produced from cadmium xanthate
3.2.1 Spectra and size distributions
The synthesis has been described recently [37]. Fig. 9 shows typical UV–visible absorption spectra of CdS particles produced from cadmium xanthate in hexadecylamine at different reaction times, corresponding to different particle sizes.
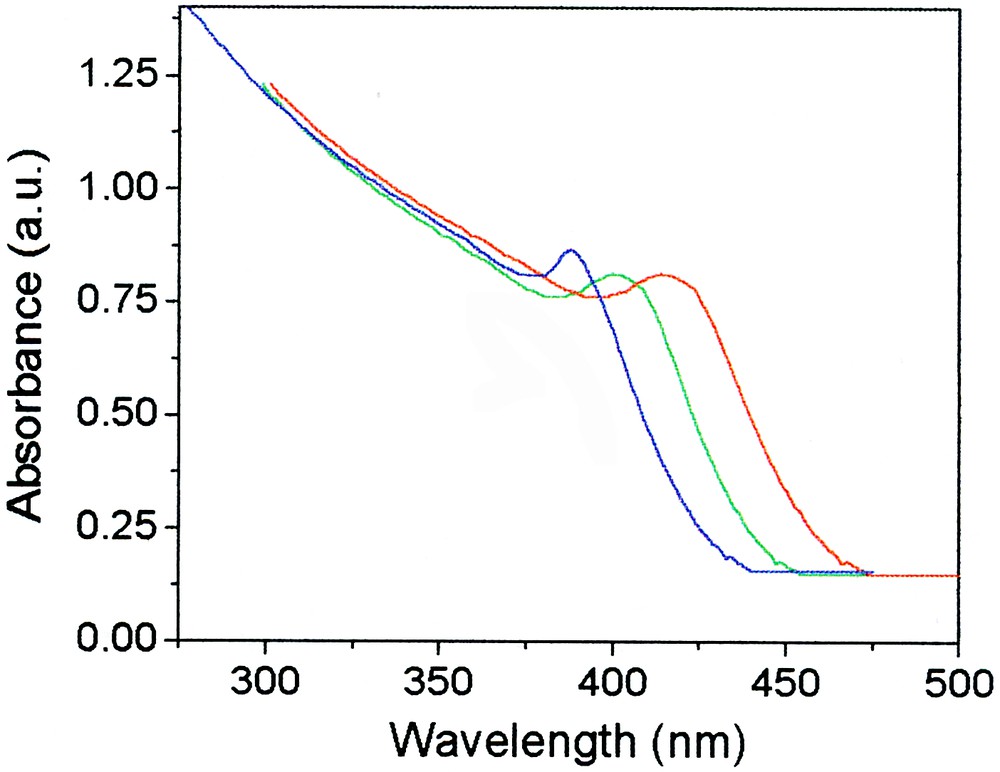
UV–visible absorption spectra of CdS particles formed from cadmium hexadecylxanthate by (from left to right) 20-, 50-, 80-min reactions.
The red shift of the spectrum with time indicates the quantum confinement in the small particles, and the clear peaks attest to a narrow size distribution. Theoretical analysis of the thresholds in the spectra (Eq. (1)) [45, 46] gives an average diameter of the particles of 3.6, 4.7, 5.5 nm (±0.4 nm). This is in full agreement with the size distribution observed in transmission electron microscopy (Fig. 10).
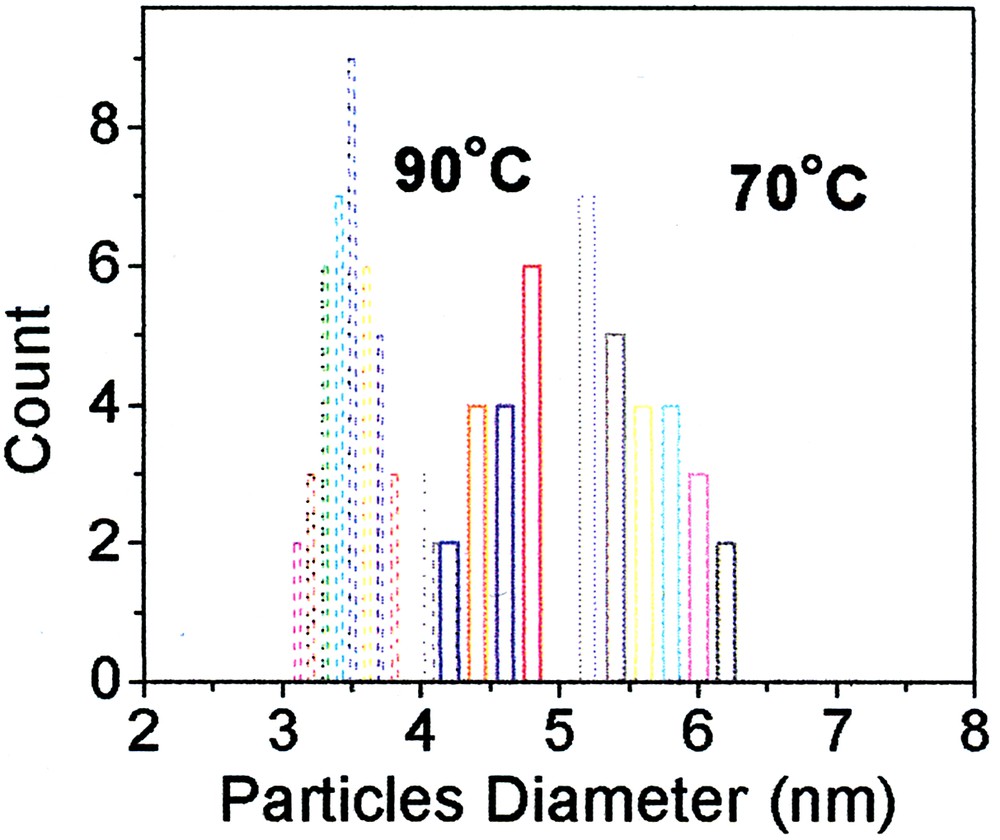
Size distribution of CdS particles produced at two temperatures, as observed in TEM.
Here Egb is the band gap of the bulk semiconductor, Egn is the band edge in the absorption spectrum, m* is the electronic effective mass and R is the radius of the particle.
The fluorescence spectra (after annealing) shown in Fig. 11 exhibit very sharp (30–35 nm FWHM) excitonic [47] emission, which is precisely tunable over a large range.
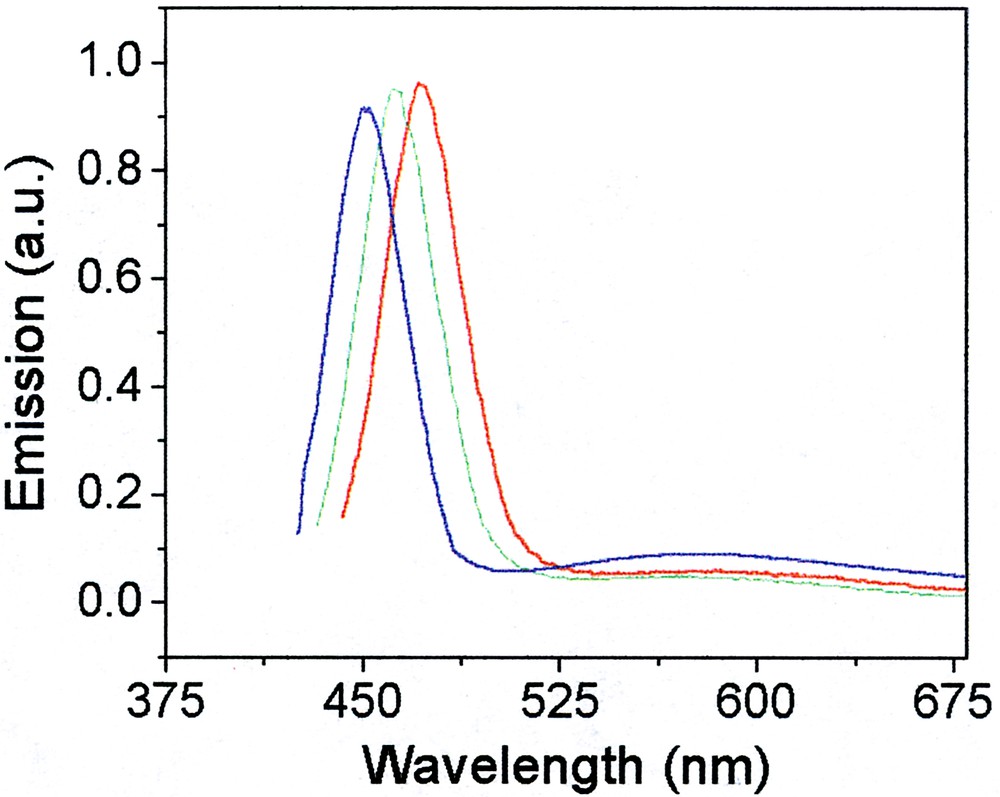
Fluorescence spectra of CdS particles formed from cadmium hexadecyl xanthate by (from left to right) 20-, 50-, 80-min reactions.
By controlling the degree of annealing, one can retain for the same particles also a broad long wavelength emission (at 520–650 nm) stemming from surface states (deep traps) [44, 47]. In fact, one can set almost any desired ratio between the two types of emissions, the excitonic and the surface fluorescence (Fig. 12). Thus one can have dual-band fluorescing particles or single-band ones.
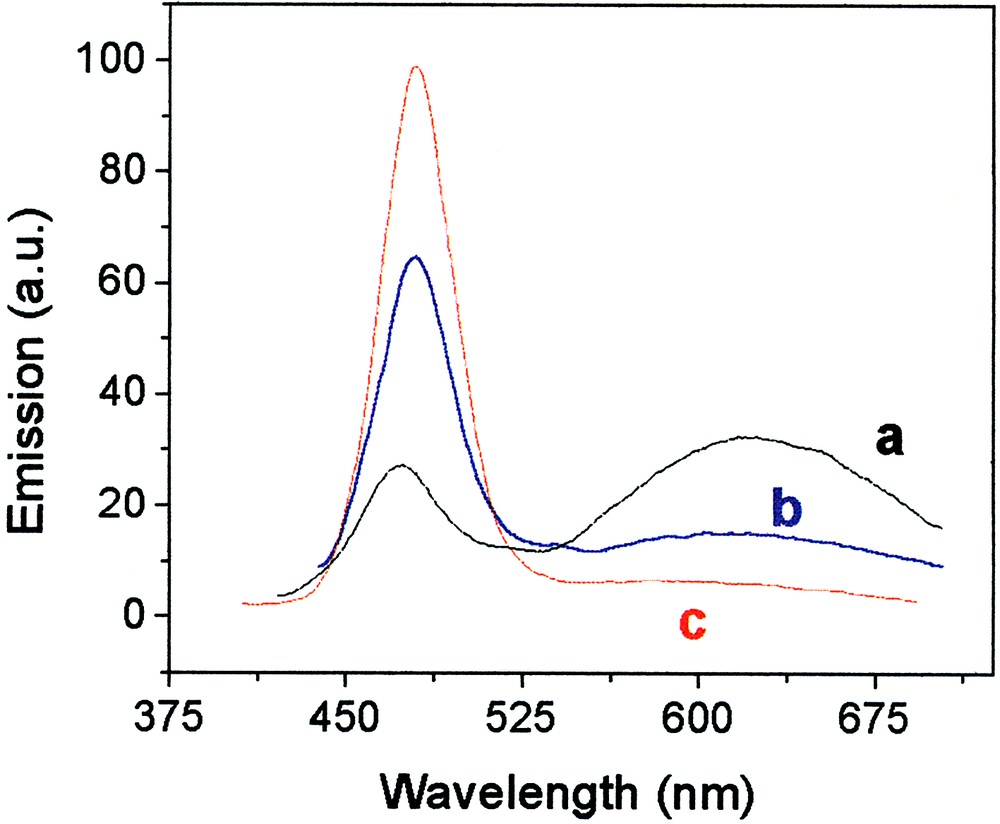
Photoluminescence spectra of CdS particles as a function of duration of annealing at 120 °C. Curves a, b and c are for annealing times are 10, 40 and 90 min, respectively.
The typical quantum yield of the narrow excitonic fluorescence of these particles is ~1–2% (excitation at 412 nm) or 2.4 % with excitation at 372 nm. Such a low quantum yield is characteristic of CdS particles [47–49]. However, and most importantly, the fluorescence of the CdS particles does not decay even after hours of continuous UV illumination. This is a big advantage of fluorescing nanoparticles over conventional fluorescence molecular probes.
Similar results to those of xanthates are obtained also for thiocarbamates and trithiocarbonates (Fig. 13). However, the carbamates require much higher temperatures in order to react (170 °C) while trithiocarbonates require intermediate reaction temperatures (140 °C). This can be taken used in single-pot methods for the production of core/shell structures as demonstrated in section 3.3.3.
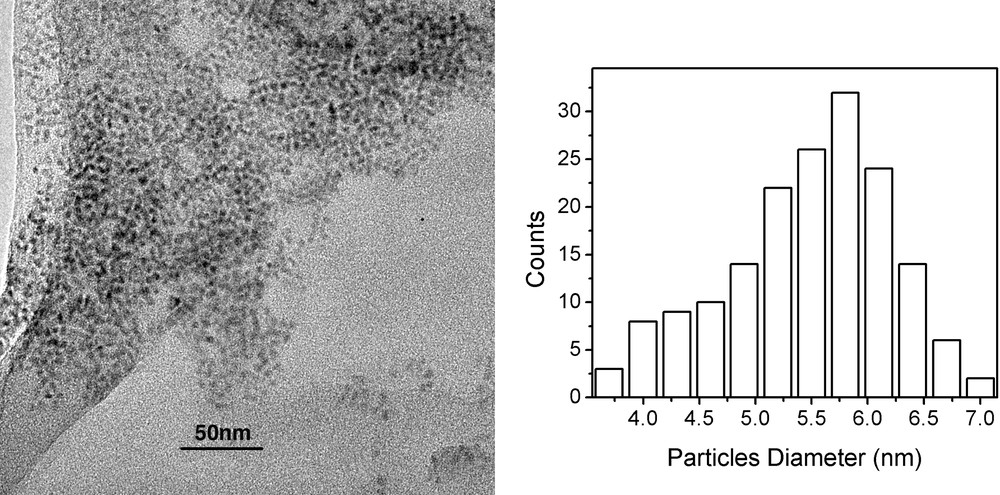
TEM (left) and size distribution histogram (right) of CdS particles formed from cadmium trithiocarbonate at 170 °C.
3.2.2 CdS particles of different shapes [50]
The versatility of our method is extended to the production of non-spherical particles, such as rods and faceted particles. With increasing concentration or by elevating the temperature above 150 °C during the synthesis or subsequent to it we obtain changes in the shape (and the corresponding spectra).
Fig. 14 shows CdS rods produced from a 1:1 weight ratio of Cd xanthate to HDA (high concentration) heated to 120 °C. Uniform crystalline rods are observed with widths of 5 nm (±1 nm) and lengths of 25 nm (±4 nm).

TEM of CdS rods formed at 120 °C with 1:1 weight ratio of cadmium hexadecylxanthate to HDA.
Faceted particles are also obtained from cadmium hexadecylxanthate in HDA at reaction or annealing temperatures above 150 °C [50].
3.3 ZnS particles and CdS/ZnS and ZnS/Cds core/shell structures
3.3.1 ZnS particles
ZnS nanoparticles can be produced in a similar fashion starting from Zn-xanthate (or the carbamates or trithiocarbonates). The temperatures required are usually somewhat lower than that required for the decomposition of the corresponding Cd salts. Fig. 15 shows the absorption spectrum of such ZnS particles.
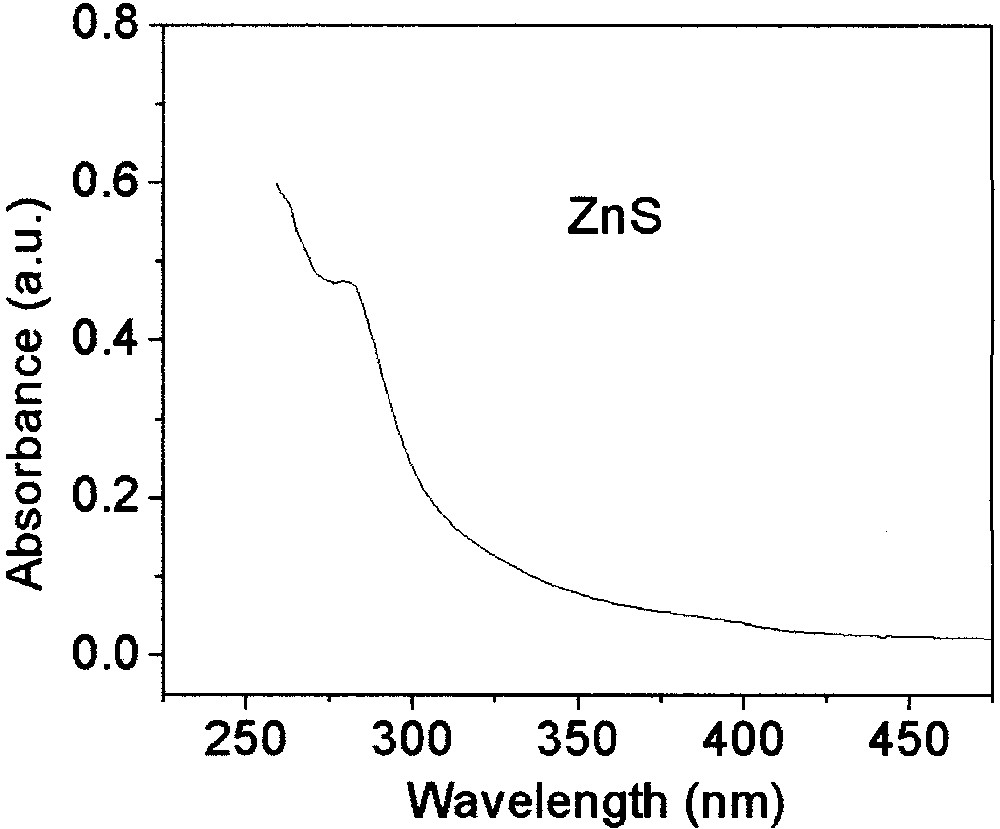
UV–visible absorption spectrum of ZnS particles synthesized from Zn-hexadecyl xanthate at 120 °C.
Once again the quantum confinement is manifested in the shift of the absorption edge compared to the bulk [51–53] with particle size. Since ZnS particles are only weakly fluorescing we focus on their use in core/shell structures.
3.3.2 CdS/ZnS core/shell particles [50]
Shelling of CdS particles produced from xanthates by ZnS is straightforward [50]. Briefly, Zn-xanthate in HDA is added to CdS particles produced from cadmium xanthate in HDA and gradually heated from 60 to 150 °C. This is repeated with a second lot of Zn-xanthate. After annealing at 170 °C, the particles are collected by flocculation with methanol at 70 °C.
The shelling of the CdS particles is evidenced from various findings. (1) A slight blue shift of the exciton emission peak from 450 nm to 436 nm is observed (Fig. 16). (2) A large increase of the integrated quantum yield of the excitonic fluorescence of the CdS particles (up to a quantum yield of 13.9 ± 0.3%), is measured. As far as we are aware, this is a record of the excitonic quantum yield of CdS particles. (3) The Zn-xanthate treated particles exhibit a significant thermal stability compared to the bare ones. The shell apparently prevents the shape transition at 150 °C discussed above. (4) A further decrease (and elimination) of the broad defect-originated long wavelength fluorescence is strongly indicative of shelling (Fig. 16).

UV–visible and PL spectra of core CdS (red) and with shelled ZnS (blue) particles synthesized at 120 °C and annealed at 170 °C.
3.3.3 ZnS particles coated with CdS [50]
ZnS/CdS core shell nanoparticles can be produced in a similar manner to the production of CdS/ZnS particles with a slightly different temperature protocol [50]. However, we also developed a novel one-pot synthesis of core/shell ZnS/CdS particles [50]. We use Cd-hexadecylthiocarbamate as the source for CdS (decomposition at ~170 °C) and Zn-hexadecylxanthate as the source of ZnS (decomposition at 70 °C). (optionally under a flow of nitrogen). The temperature of a mixture of these reagents in HDA is gradually increased from below 60 up to 200 °C. First, the Zn xanthate reacts and then the ZnS particles become shelled by CdS. The reaction is monitored by measuring the fluorescence peak position.
These ZnS/CdS particles can be used as nucleation seeds for the production of yet more complex structures, such as ZnS/CdS/ZnS. Core-shell-shell particles, or quantum-dot-quantum-wells, have been of considerable recent interest [54–56].
Fig. 17 shows a comparison of the emission from ZnS/CdS and ZnS/CdS/ZnS particles (excitation at 370 nm).
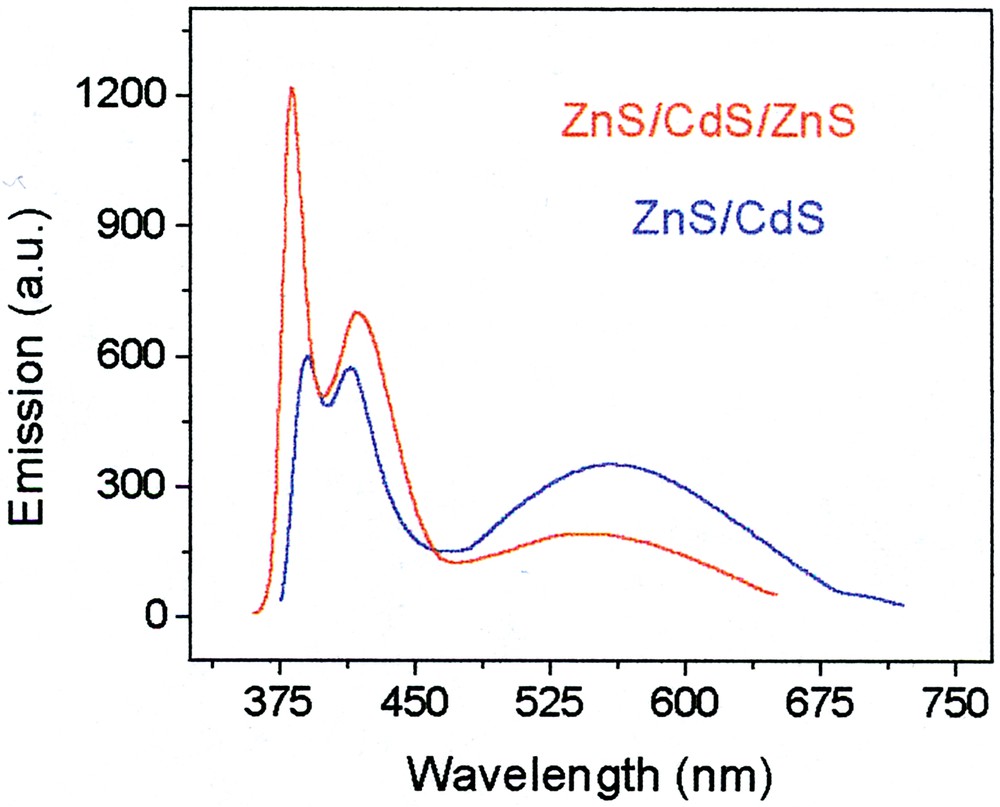
Photoluminescence spectra of ZnS/CdS (blue) and ZnS/CdS/ZnS (red) particles.
The ZnS/CdS particle exhibits three emission bands. The very broad one centered at~545 nm is the familiar CdS surface defect (deep trap) fluorescence. Next, the narrow band at ~410 nm, about 30–40 nm wide is the excitonic transition. It appears at an unusually low position, probably because of the narrowness of the (perhaps incomplete) CdS shell. A third extremely narrow peak (~10 nm FWHM) appears at very high energies, 380 nm. Its origin is not yet determined.
Upon adding the third cladding ZnS layer, the surface trap fluorescence is quenched to a significant degree. The 410-nm excitonic emission shifts slightly to longer wavelengths and gains some intensity. The short wavelength band also becomes more intense (and may be blue-shifted).
3.4 Sulfide particles of other metals
As stated above, our method is very general and versatile. It can be easily applied to many other metals. We demonstrate this by one example.
Fig. 18 shows micrographs of PbS particles produced from hexadecyl xanthate at room temperature. The particles are highly crystalline and mostly are octagonal in shape.

TEM micrograph of PbS particles synthesized at room temperature from lead hexadecylxanthate
One of the main interesting features is the rapid formation of PbS and HgS particles at room temperature. In fact, we need to cool down the reaction solution in order to slow down the reaction and obtain individual nanoparticles.
4 Conclusions and future developments
We have discussed and demonstrated the use of xanthates (and related compounds) as versatile, convenient and general reagents in colloid science. Xanthates have some unique properties stemming mostly from a delicate balance between inertness and chemical and thermal instability. Thus, on the one hand, they serve as efficient capping agents for a variety of metal nanoparticles, that stabilize them and protect them against chemical ‘attack’, corrosion, for instance. On the other hand, they decomposed by heat and thus can be applied as disposable agents. This thermal (chemical) instability comes in handy when using xanthates as precursors in the synthesis of controlled (and apparently crystalline) sulfide nanoparticles. Xanthates can be used as single precursors in the form of their metal salts and can be applied in a single-pot manner, even when producing complex systems, such as core/shell and core/shell/shell particles.
Another attractive feature of xanthates is their relative higher solubility in polar liquids, compared to thiols, which are so useful in surface and colloid science.
While thiols were first developed and studied in the context of self-assembly and only later were applied to nanoparticles, with xanthates we seem to go in the opposite direction. We have demonstrated their usefulness for particles, and though we did not discuss it in this mini-review, we believe that xanthates (and thiocarbamates, trithiocarbonates, etc) should be highly effective agents in self-assembly and the modification of solid (metal-containing) surfaces. Xanthates readily adsorb on such substrates [28–31]. Depending on the functionality terminating the chain we can then impart to these systems a variety of properties and functions, just as done with thiols (carboxylates, amines and, on glass, silanes). However, similar to the silane case, care needs to be exercised to prevent reaction between the functional end and the xanthate side of the molecules. For instance, the corresponding thioesters can be utilized as protective groups.
Furthermore, if one uses the xanthate end as the functional terminus (adsorbing the molecule to a surface via a silane, thiol, carboxylate or amine group) new possibilities open up. One such possibility is carrying out the Chugaev reaction [24] on the surface and thus coating the surface with olefinic moieties. In a similar fashion surface metal sulfides can form by adsorbing metal ions to a xanthate layer followed by heating.
In summary, we suggest xanthates, thiocarbamates, trithiocarbonates and related compounds as versatile and useful new tools in the growing arsenal of nanotechnology, surface science and particle chemistry.
Acknowledgements
We acknowledge the original contribution of P.D. Sawant, O. Tzhayik and J.T. Klug to the work on metal colloids, and the helpful discussions with B. Katz. Partial support of Infrastructure Program of the Israel Ministry of Science is acknowledged.