

1 Introduction
In recent years, the elaboration of hybrid materials by the combination of organic polymerization and inorganic polycondensation has attracted much interest because of the unusual properties that may arise from the incorporation of inorganic components into organic polymers [1–20]. These hybrids can be elaborated in a variety of ways. Pioneering works in this field have been done by Schmidt and Mckenzie, who synthesized hybrid glasses through the impregnation of silica gels with organic monomers followed by in-situ free radical polymerization [1–2]. Inversely, Landry and co-workers reported on the formation of molecular composites by the in-situ polycondensation reaction of metal alkoxides in various organic polymers through the sol–gel process [3–5]. The so-produced sol–gel derived composite materials were optically transparent and displayed enhanced mechanical properties [6]. In order to synthesize homogeneous polymer hybrids and to improve the chemical bonding between the organic and inorganic components, organic polymers carrying functional groups such as poly(oxazoline) [6], poly(vinyl alcohol) [6], poly(acrylamide) [7], poly(dimethylsiloxane) [8], and various trialkoxysilane-terminated polymers [6,9–11] have been involved in the elaboration of the hybrid glasses. Alternatively, mixtures of vinyl monomers and sol–gel precursors have been used to afford organic/inorganic simultaneous interpenetrating networks wherein both inorganic glass and polymer formation occur concurrently [12–16]. The properties of this last class of hybrids can be significantly improved by using coupling agent molecules reactive in the free radical process, as for instance organo alkoxysilanes bearing a methacrylate end group. The silane molecules were shown to improve the adhesion between the organic and inorganic components, and efficiently control phase separation in the resulting hybrid materials.
Although there has been a huge number of works in the field of organic/inorganic hybrids since the 1980s, surprisingly the combination of free radical polymerization and metal alkoxides polycondensation reactions has been scarcely performed in multiphase systems, e.g., through emulsion or miniemulsion polymerizations [17–23]. Emulsion polymers, however, are widely used in industry and can find applications in a variety of domains including water-borne adhesive, paint and coating formulations. In coating applications, the final properties of the polymer film strongly depend on the film formation process and can be significantly enhanced by incorporating functional moieties capable of undergoing chemical or physical cross-linking either during polymerization or after synthesis during film formation [24–26]. To this goal, organo alkoxysilane derivatives appear to be versatile compounds to provide self-cross-linking ability to water-borne copolymer films, and therefore enhance the cohesive strength and integrity of the coating.
Following our previous works in this field [19–21], we describe in this paper the elaboration of hybrid latexes by reacting simultaneously a polymerizable organosilane derivative: 3-trimethoxysilyl propyl methacrylate (MPS), and conventional styrene (Styr.) and n-butyl acrylate (BuA) monomers in emulsion polymerization. When MPS is introduced in the organic monomers mixture, two sets of chemical reactions can occur simultaneously. First, the silane monomer can be incorporated into the polymer chains by conventional free radical copolymerization reactions as depicted in Scheme 1 , and, second, the trimethoxysilyl groups in the copolymers can undergo hydrolysis/polycondensation reactions resulting in cross-linking of the hybrid film as schematically represented in Scheme 2 . In addition, the introduction of unreacted residual alkoxy silyl and silanol groups into the hybrid copolymers makes it possible to produce self-curable films which can be post cross-linked upon increasing pH and/or temperature. We report in the following on the formation and characterization of such film-forming hybrid latexes. The emphasis is put in the present work on the microstructure and the mechanical properties of the composite film materials.
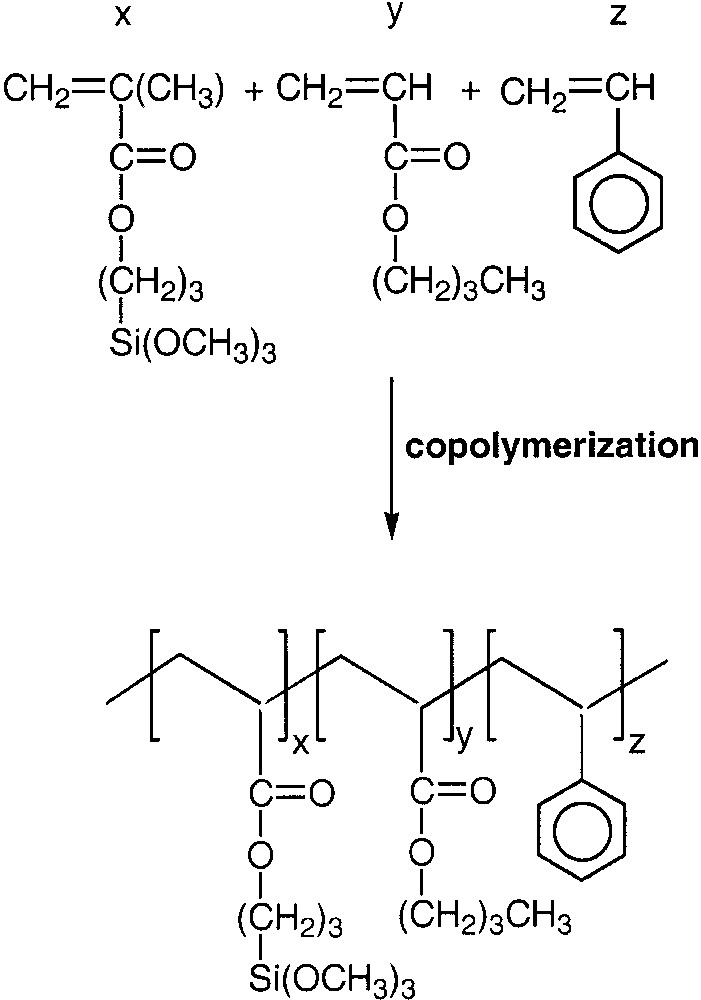
Reaction scheme for the copolymerization reaction of MPS, styrene and n-butyl acrylate monomers.

Reaction scheme for the hydrolysis/condensation reactions of the hybrid copolymers into the polymer latexes.
2 Experimental section
2.1 Materials
The monomers, styrene and butyl acrylate (from Aldrich), were distilled in vacuum before use. The initiator, potassium persulphate (KPS, Acros Organics) and the surfactant, sodium dodecyl sulphate (SDS, Aldrich), were used as supplied. 3-Trimethoxysilyl propyl methacrylate (MPS, Aldrich) was used without purification. The water was deionized on mixed bed resins before use.
2.2 Synthesis
Unless stated otherwise, the emulsion polymerization reactions were carried out in batch at 70 °C for 8 h under nitrogen atmosphere. The 300-ml glass reactor fitted with a condenser was charged with the anionic surfactant (SDS) and deionized water. The pH of the suspension was fixed at pH 7 by adding equivalent quantities of dihydrogenophosphate and dihydrogenocarbonate from Acros Organics (typically 0.1 g of each per 100 g of water). Degassing was carried out for around half an hour under nitrogen atmosphere and gentle stirring before increasing the temperature up to 70 °C. Then, the monomers and the initiator dissolved in 10 ml of deionized water were added at once to start polymerization. A typical recipe is as follows: monomers 10 g, KPS 0.1 g (0,37 mmol), SDS 0.2 g (0,69 mmol), buffer 0.2 g, water 100 g. The (co)monomers feed composition and the samples code are given in Tables 1 and 2.
Feed compositions and samples code for the emulsion copolymerization reactions of 3-trimethoxysilyl propyl methacrylate (MPS) and n-butyl acrylate (BuA) monomers
| Feed composition | ||
| Samples code | MPS (g l–1) | BuA (g l–1) |
| P(BuA) | 0 | 99.7 |
| P(BuA-co-MPS) (5%) | 5.1 | 94.9 |
| P(BuA-co-MPS) (10%) | 10.1 | 89.9 |
| P(BuA-co-MPS) (15%) | 15.3 | 84.8 |
| P(BuA-co-MPS) (20%) | 20.1 | 80.1 |
| P(BuA-co-MPS) (30%) | 30.2 | 72.2 |
Feed compositions and samples code for the emulsion terpolymerization reactions of 3-trimethoxysilyl propyl methacrylate (MPS), n-butyl acrylate (BuA) and styrene (Styr) monomers
| Feed composition | |||||
| Samples code | MPS (g l–1) | Styrene (g l–1) | BuA (g l–1) | Styr/BuA a | Styr/BuA b |
| P(Styr-co-BuA) | 0 | 37 | 63 | 37.0/63.0 | 36.0/64.0 |
| P(Styr-co-BuA-co-MPS) (5%) | 5 | 35.0 | 60.0 | 36.8/63.2 | 35.9/63.1 |
| P(Styr-co-BuA-co-MPS) (10%) | 10 | 32.9 | 57.0 | 36.6/63.4 | 35.7/64.3 |
| P(Styr-co-BuA-co-MPS) (15%) | 15 | 31.4 | 53.6 | 37.0/63.0 | 36.0/63.0 |
| P(Styr-co-BuA-co-MPS) (20%) | 20 | 30.0 | 50.0 | 37.5/62.5 | 36.6/63.4 |
| P(Styr-co-BuA-co-MPS) (30%) | 30 | 26.1 | 44.1 | 37.2/62.8 | 36.3/63.7 |
| P(Styr-co-BuA-co-MPS) (40%) | 40 | 22.0 | 38.1 | 36.6/63.4 | 35.7/64.3 |
2.3 Films formation
The hybrid films were prepared by casting the polymer latexes in Teflon dishes and evaporating water at 70 °C for one night until the formation of homogeneous solid films. Samples with a controlled thickness, ranging between 0.2 and 0.4 mm, were cut from the films and stored at ambient temperature before use.
3 Characterizations
3.1 Monomer conversions and copolymer particles composition
The monomer-to-polymer conversions were determined gravimetrically by drying a known volume of the aqueous suspension at 70 °C, and weighing the residue. We assumed that the polycondensation reaction of the silane molecule gave polymers with the following stoichiometry: RSiO1,5. The volatile species were alternatively quantified by gas chromatography (Hewlett-Packard 5890 Series II) by diluting the latex suspension in a known amount of THF used as an internal standard. The copolymer composition was determined by elemental analysis from the silicon content using Eq. (1) below:
| (1) |
3.2 Spectroscopic analyses
Infrared spectra were recorded using a Nicolet FTIR 460 spectrometer on thin films obtained by coalescence of a 1-wt% diluted suspension of the hybrid latexes. 29Si and 13C solid state NMR were performed on a Bruker DSX-300 spectrometer operating at 59.63 and 75.47 MHz, respectively by use of cross-polarization from proton. The contact time was 5 ms, the recycle delay 1 s and the spinning rate 10 kHz. The 29Si NMR spectra have been simulated using the DMFIT program for quantitative analysis [27].
3.3 Particles size and morphology
The intensity-average hydrodynamic diameter, Dp, of the latex particles was determined by dynamic light scattering (DLS) using a Malvern Autosizer Lo-C instrument. Transmission electron microscopy analysis (TEM) was performed with a Philips CM10 electron microscope operating at 80 kV. In a typical experiment, one highly diluted drop of the colloidal dispersion, containing approximately 0.01 wt% of solid, was put on a carbon film supported by a copper grid. Negative staining was performed by adding one drop of an aqueous solution of 1-wt% uranyl acetate on the copper grid, and allowed to air dry before observation.
3.4 Dynamic mechanical analysis
Dynamic mechanical analysis (DMA) measurements were performed in torsion mode with a homemade inverted pendulum. Measurements were performed at a fixed frequency (1 Hz) from –180 to 100 °C with a heating rate of 1 K min–1. The imposed deformation was about 10–4, which ascertains that the measurements were in the linear viscoelastic region. Sample dimensions were around 4 × 2 × 0.3 mm3. The set-up provides the storage and the loss components of the complex shear modulus (G′ and G" respectively), and the ratio of these two components i.e. tanδ =G"/G′.
4 Results and discussion
4.1 Synthesis of the hybrid copolymer and terpolymer latexes
4.1.1 Particle sizes and monomer conversions
P(BuA-co-MPS) copolymer and P(Styr-co-BuA-co-MPS) terpolymer latexes containing different silane contents were prepared in batch through emulsion polymerization according to the recipes given in Tables 1 and 2. Based on our previous works in this field [19–21], the latexes were stabilized using an anionic surfactant and the suspension pH was adjusted to 7. These experimental conditions were chosen such as to ensure a good colloidal stability of the hybrid copolymer latexes and to control hydrolysis and condensation reactions of the silane units. Stable latexes were indeed obtained without the formation of coagulum whatever the silane feed composition. Fig. 1 shows the evolution of particles size as a function of the silane content in the feed. Particles diameter decreased from around 80 nm to 50 nm with increasing the MPS feed concentration from 0 to 20 wt%, for both the copolymer and the terpolymer latexes.

Hybrid polymer particles diameter as a function of the silane content in the feed - -: P(Styr-co-BuA-co-MPS); - • -: P(BuA-co-MPS)..
The decrease in particles size with increasing silane content can be attributed to the incorporation of silanol groups into the hybrid particles, which presence contributes to increased stability of the colloid. Indeed, as shown in a previous work, increasing the MPS concentration leads to an increase of the silanol groups’ density [20]. The amount of surface charges thus increases, while concurrently less surfactant is adsorbed on the surface (the area per surfactant molecules increases). Fig. 2 shows a typical TEM micrograph of P(BuA-co-MPS) copolymer latex particles with 20 wt% MPS. The particles have a narrow size distribution indicating that no extensive renucleation occurred in the course of polymerization. However, although they are not discernable by TEM analysis, the formation of small polyorganosiloxane clusters in the aqueous phase through condensation reactions cannot be completely ruled out at the present stage. Such nano-sized clusters might contribute to a decrease of the average particles size.

TEM micrograph of hybrid copolymer latex particles with 20% silane content.
Both gravimetric measurements and gas chromatography analysis gave evidence of full incorporation of the silane molecules in the copolymers up to 20-wt% silane content in the feed for the P(BuA-co-MPS) copolymers, and up to 30wt% for the P(Styr-co-BuA-co-MPS) terpolymers. For higher concentrations, we observed a decrease in the overall solid content and a limitation of monomer conversions for both series of samples, while concurrently, infrared spectroscopy and 13C NMR analysis revealed the presence of unreacted methacrylate in the P(BuA-co-MPS) copolymers with the appearance of a signal at 1637 cm–1 and peaks at 138 and 122 ppm, respectively. All these results indicate that the silane units have been incorporated in the polymer chains through hydrolysis and subsequent condensation reaction (cross-linking) with previously attached silanol groups without copolymerizing. Similar results have been reported by Marcu et al. during the miniemulsion copolymerization reaction of n-butyl acrylate and vinyltriethoxysilane monomers, and were attributed to the low reactivity of the silane moiety towards the PBuA macroradicals [22]. The reactivity ratios for the copolymerization reaction of styrene (monomer 1) and MPS (monomer 2) were reported to be r1 = 0.45 and r2 = 0.9, respectively [28], while those for BuA (M1) and MPS (M2) were estimated to be r1 = 0.005 and r2 = 0.007, respectively. These values indicate that copolymerization is promoted and that MPS units should be randomly incorporated into the copolymer chains. Therefore, we suspect in the present work the formation of small water insoluble cyclic oligomers that can no longer diffuse through the aqueous phase and copolymerize. Although it is still unclear to us, the decrease in the overall monomer conversion with increasing MPS concentration may also be due to monomer entrapment within the inorganic network as suggested in earlier studies. The hybrid copolymers were further characterized by elemental analysis (Table 3). The copolymer compositions, expressed by the fraction in weight of MPS in the copolymers, were found to be in good agreement with the (co)monomers feed compositions except for large silane content in agreement with the previous discussion.
Feed and copolymer compositions determined by elemental analysis of the hybrid film materials
| Feed composition | Copolymer composition | ||
| Samples code | MPS (wt%)a | Si (wt%) | MPS (wt%)b |
| P(Styr-co-BuA-co-MPS) (10%) | 10 | 1.2 | 10.3 |
| P(BuA-co-MPS) (5%) | 5.1 | 0.6 | 5.1 |
| P(BuA-co-MPS) (10%) | 10.1 | 1.2 | 10.3 |
| P(BuA-co-MPS) (30%) | 30.2 | 4.9 | 42.1 |
4.1.2 Copolymers microstructure
The copolymers microstructure was characterized by solid-state NMR and FTIR spectroscopies. Typical FTIR spectra of hybrid copolymer and terpolymer films along with spectra of the organic polymers alone, are shown in Figs. 3 and 4 , respectively as a function of the silane feed composition.
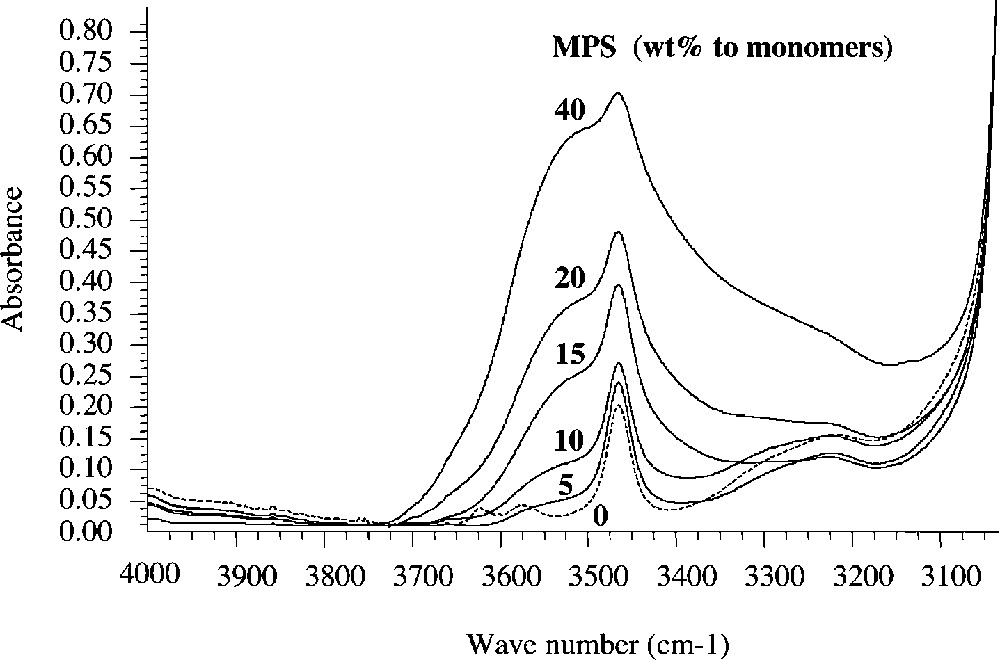
FT–IR spectra of the P(BuA-co-MPS) copolymers with increasing silane contents in the feed.
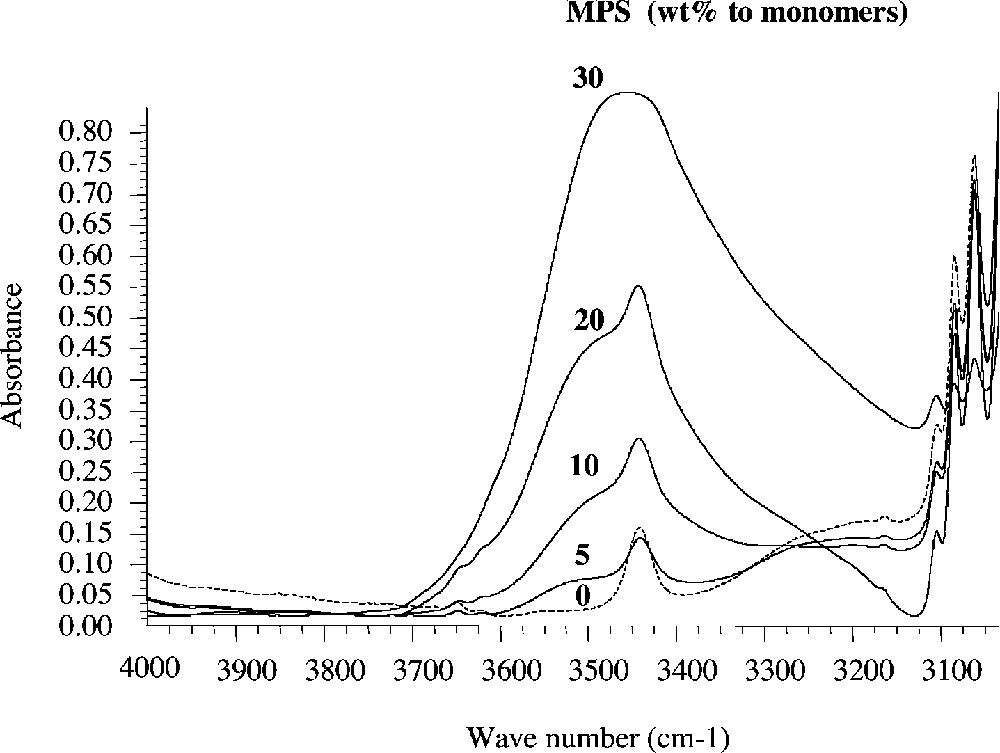
FT–IR spectra of the P(Styr-co-BuA-co-MPS) copolymers with increasing silane contents in the feed.
The two series of spectra exhibit all the characteristic bands of the PBuA and P(Styr-co-BuA) homopolymer and copolymer components, along with signals characteristics of the siloxane network. The presence of hydroxyl groups is evidenced by the appearance of a broad absorption peak at 3450 cm–1, which intensity increased with increasing silane content.
The 13C NMR spectra of the latex films also support the hybrid structure of the copolymers. The signals at 137.4 and 124.7 ppm, assigned to the MPS double bonds, have disappeared while peaks characteristic of the organic polymers can be clearly identified at positions corresponding to the chemical shifts of the polymer matrix used as reference. The condensation reactions leading to the formation of Si–O–Si bridges (Scheme 2) was followed by solid-state 29Si NMR spectroscopy. The different species are called according to the conventional Tn notation where T designates a trifunctional unit and n is the number of bridging O atoms surrounding the silicon atoms. Three main peak regions, corresponding to T1, T2 and T3 units can be identified in the NMR spectra, and the extent of cross-linking can be estimated accordingly by determining the area under each of the three peaks. The percentage of the different Tn species is reported in Table 4 for a series of selected samples. To afford reliable data, only the hybrid copolymers that contained a significant amount of MPS (up to 5 wt%), and those that were fully converted were analysed by 29Si NMR.
Results of the deconvolution of the 29Si NMR spectra of a series of copolymer and terpolymer latexes
| Entry | Samples code | T0 | T1 | T2 | T3 | |
| Terpolymer latexes | 1 | P(Styr-co-BuA-co-MPS) (10%) | 39 | 20 | 31 | 10 |
| 2 | P(Styr-co-BuA-co-MPS) (20%) | 6 | 18 | 57 | 19 | |
| 3 | P(Styr-co-BuA-co-MPS) (20%) * | 4 | 36 | 34 | 26 | |
| Copolymer latexes | 4 | P(BuA-co-MPS) (20%) | 0 | 0 | 48 | 52 |
| 5 | P(BuA-co-MPS) (20%) * | 0 | 20 | 50 | 30 |
The results in Table 4 indicate that the cross-linking reaction is dependent on the silane concentration. The larger the silane content in the feed, the higher the condensation degree of the silica-based network (entries 1 and 2). This can be attributed to the increase of the inorganic condensation rate with increasing the MPS concentration. Premature hydrolysis and cross-linking can be partially avoided nevertheless, for both the terpolymer (entry 3) and the copolymer (entry 5) film materials, using a semi-continuous addition profile of the monomers. In a typical experiment, a mixture of styrene, n-butyl acrylate and MPS is added drop wise to a latex seed suspension at the feeding rate of 18 g l–1 h–1. In these conditions, the instantaneous MPS concentration in the water phase is low, and the condensation rate is significantly decreased as attested by NMR analysis. The fractions of T0 and T1 species corresponding to un-completely condensed silane units increased, while the peaks area under the T2 and T3 signals decreased in agreement with previous reports in this field [29]. The organic polymerization also has an influence on the extent of cross-linking, as already demonstrated in a previous work [21]. The latexes obtained by reaction of styrene, n-butyl acrylate and MPS (entry 2) exhibited less premature cross-linking than the one produced by copolymerizing MPS and BuA (entry 4). This is presumably due to the high reactivity of MPS towards the polystyryl radicals, which promotes organo alkoxysilane incorporation into the copolymer chains and minimizes their prolonged contact with water [28]. It is worth noticing however that the terpolymer film material with 20wt% silane content contains only a small fraction (6%) of un-condensed monomeric MPS units (T0 species), and that a high proportion of the silicon atoms (>76%) has been involved in cross-linking reactions.
4.2 Films formation
The films produced from the hybrid latexes were shown to display good optical transparency and homogeneity for up to 20wt% MPS. They were all insoluble in good solvents for PBuA and P(Styr-co-BuA), such as toluene and THF, indicating that the polymer chains were homogeneously cross-linked thorough the film in agreement with the above characterizations. Fig. 5 shows the photograph of a representative transparent composite film with 10wt% silane content.

Photograph of transparent hybrid terpolymer latex film containing 10-wt% MPS.
4.3 Mechanical behaviour
Fig. 6 shows the dynamic viscoelastic properties versus temperature for the hybrid copolymers and terpolymers with MPS contents of 0, 10 and 20 wt%, respectively. For all the samples, the modulus in the glassy plateau has been normalized to a value of 1 GPa.

Dynamic viscoelastic properties of the hybrid films as a function of the MPS content in the feed. Isochronal (frequency = 1 Hz) temperature dependence of G′ (a, b) and tan δ (c, d).
The copolymer samples, PBuA and P(Styr-co-BuA), display typical behaviour of amorphous thermoplastic polymers. At low temperature, i.e. in the glassy state, the modulus remains roughly constant; actually it slightly decreases with increasing temperature. Then, a rapid decrease in the elastic part of the modulus is observed while tan δ passes through a maximum. This relaxation phenomenon is associated to an energy dissipation involving cooperative motions of long chain sequences, in relation with the glass-transition phenomenon. It takes place at –33 °C for PBuA and 17°C for P(Styr-co-BuA) as given by the maximum of tan δ. Then, the elastic part of the modulus G′ reaches a plateau (rubbery modulus) around 0.1 and 0.2 Mpa for PBuA and P(Styr-co-BuA), respectively. At higher temperature, the increase of tan δ indicates the beginning of macromolecule flow, as expected in a thermoplastic polymer.
In the case of the hybrid specimens, it can be first observed that the main mechanical relaxation is shifted towards higher temperatures. In the case of PBuA, the maximum of tan δ increases from –33 to –15 °C with an addition of 10% of MPS, and to –10 °C with an addition of 20% of MPS. In the same time, the dynamic modulus in the rubbery plateau, G′, increased drastically. For instance, it reaches a value around 10 MPa with 20% of MPS that corresponds to an increase of two decades. Similar trends are observed in the case of the P(Styr-co-BuA) copolymer-based hybrids. With 10% of MPS, the shift of the maximum of the tan δ peak is 17 °C and it reaches 40 °C with 20% of MPS. The observed increase of rubbery modulus is once again around two decades. Moreover, in the studied temperature range, the increase of tan δ due to macromolecule flow is no more observed: all hybrid specimens behave like cross-linked polymers. All these evolutions are consistent with the idea of a co-continuous rigid network due to the presence of MPS in the sample as supported by the above reported micro structural investigation. This assumption might be confirmed by further morphological characterizations of the cross-linking process and inorganic network formation within the hybrid films. Different techniques could provide useful information as for instance small-angle X-ray scattering measurements (SAXS) or atomic force microscopy (AFM).
5 Conclusion
A series of hybrid copolymer and terpolymer latexes has been prepared by reacting simultaneously MPS, styrene and n-butyl acrylate in emulsion polymerization. The hydrolysis/polycondensation reaction of the silane molecule was followed by 29Si solid-state NMR spectroscopy, and the whole synthetic procedure was characterized by gas chromatography, gravimetric analysis, FTIR and 13C NMR spectroscopy. While, a limiting conversion effect was observed for MPS feed concentrations higher than 20–30wt% based on monomers, full incorporation was reported for lower silane contents. Homogeneous and optically transparent films were produced by coalescence of the latexes. Although we observed small differences in cross-linking ability of the latex films depending on the monomers feed composition and the silane addition profile, both the copolymer and terpolymer hybrids showed the occurrence of predominant cross-linking reactions which result in the production of composite films with improved mechanical properties. An increase of the elastic part of the modulus in the rubbery plateau by a factor of two decades is observed with only 20% of MPS. Such an increase is probably due to the presence of a continuous rigid network involving MPS units.
Acknowledgements
The authors thank Christian Novat from the ‘Laboratoire de chimie et procédés de polymérisation’ (Villeurbanne) for his great help in doing the TEM experiments.