

1 Introduction
Recent years have witnessed rejuvenation in the research of free radical polymerization with the development of living radical polymerization (LRP) techniques. Unlike conventional radical polymerization, in which growing radical chains have a lifetime of only ~0.1–1 s before they undergo irreversible radical–radical termination, in LRP the chains are reversibly deactivated and continue to add monomer throughout the polymerization. Irreversible termination does occur to a limited extent, but most of the chains remain ‘living', thereby enabling the synthesis of polymers with narrower molecular weight distributions and better control of the microstructure in comparison to conventional radical polymerization, making them suitable for producing block, star and comb polymers. There are three important types of LRP systems, distinguished by the nature of the species used to reversibly terminate the growing radicals; nitroxide-mediated radical polymerization (NMRP), atom transfer radical polymerization (ATRP) and reversible addition fragmentation transfer (RAFT). While research on these systems initially focused on homogeneous systems (bulk, solution polymerization), there has been considerable activity in the past few years on adapting LRP systems to heterogeneous polymerizations, especially emulsion and miniemulsion polymerization.
The well-known advantages offered by emulsion/miniemulsion polymerization include polymer reaction engineering concerns such as improved heat transfer and ease of mixing in comparison to viscous bulk/solution polymerizations, environmental advantages because water is used as the continuous phase, and the extensive experience industry has with emulsion polymerization. Unfortunately, another key advantage of emulsion polymerization, that of increased reaction rates and molecular weight due to radical compartmentalization, is not realized with NMRP or ATRP systems, although RAFT systems do retain the advantage of increased rate. Two recent reviews of LRP in heterogeneous systems have recently appeared [1,2], but there have been significant advances since their publication. This paper reviews recent progress is using nitroxide-mediated polymerization with emulsion or miniemulsion polymerization. Results from various research groups, including our own, are discussed and analyzed to present an integrated portrait of the current status of NMRP in emulsion/miniemulsion polymerization and identify the most pressing concerns, issues, and opportunities.
2 Nitroxide-mediated radical polymerization
Nitroxide-mediated radical polymerization, also known as Stable Free Radical Polymerization (SFRP), employs a stable nitroxyl such as TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) to reversibly terminate growing polymer chains (Scheme 1 ), yielding a dormant chain. The equilibrium is shifted strongly toward the dormant species so that the active (propagating) radical concentration is lower than in conventional radical polymerization. Under typical reaction conditions, a dormant chain may be activated every ~102–103 seconds on average, and add ~0–5 monomer units before deactivation occurs. The deactivation step is fast but slightly slower than diffusion-controlled rates, and leads to an average activation period of ~10–4–10–3 s. Since propagation is first order with respect to radical concentration, while irreversible biradical termination is second order, the lower radical concentration results in a significantly reduced termination rate that preserves the living character of the chains. Irreversible termination, which leads to dead chains and broadening of the molecular weight distribution, cannot be eliminated but under appropriately chosen conditions its rate is minimized. Irreversible termination also leads to higher nitroxide concentration, which forces the equilibrium to shift toward the dormant state, thereby lowering the active radical concentration and reaction rate. To minimize the breadth of the molecular weight distribution, conditions are selected so that all chains are initiated within minutes of the start of reaction. Initiation may be done using either bicomponent systems (conventional free radical initiator and nitroxide are added to the reaction mixture) or unimolecular systems (unimers) consisting of an alkoxyamine that dissociates upon heating to give a nitroxide and an active radical that can add monomer. While bicomponent systems are simpler in that unimer synthesis is avoided, using unimers provides better control of the number of polymer chains and therefore better predictability of molecular weight.
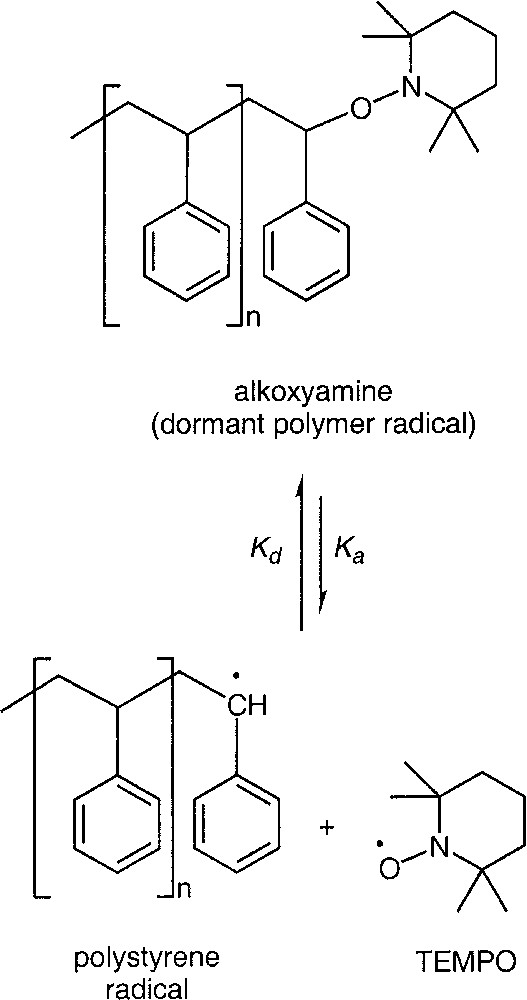
Reversible deactivation of polymeric radical by TEMPO.
The equilibrium between active and dormant chains is sensitive to temperature, and temperatures in the range 115–135 °C are necessary to achieve reasonable reaction rates using TEMPO and many other nitroxides. The acyclic phosphonylated nitroxide SG1 (N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) has a larger equilibrium constant than most nitroxides and can be used at temperatures as low as ~90 °C [3]. NMRP has proven useful in polymerization of styrenic monomers and the copolymerization of styrenics with some monomers. However homopolymerization of acrylates is often difficult, and is sensitive to the structure of the nitroxide. Success has not been achieved with methacrylates due to a rapid disproportionation reaction that involves β–H abstraction from the polymeric radical by the nitroxide to give dead, terminally unsaturated polymer and the corresponding hydroxylamine. Newer alkoxyamines have been shown to be effective for a variety of monomers, including 1,3-dienes, acrylates, acrylamides and acrylonitrile [4,5]. Monomers that do not easily autopolymerize are more difficult to polymerize with nitroxides, because the existence of biradical termination causes a gradual increase in the free nitroxide concentration until the polymerization is completely suppressed. Thermal initiation of styrene provides a source of radicals to consume the excess nitroxide concentration, thus allowing reasonable reaction rates. Mayo [6] first proposed the mechanism shown in Scheme 2 . Thermally initiated radicals can result in increasing polydispersity, however, they provide a means of mitigating the effects of nitroxide accumulation. Some thermal initiation is thus beneficial for most nitroxide-mediated polymerizations. Scheme 3 shows the important reactions and their rate expressions in NMRP.
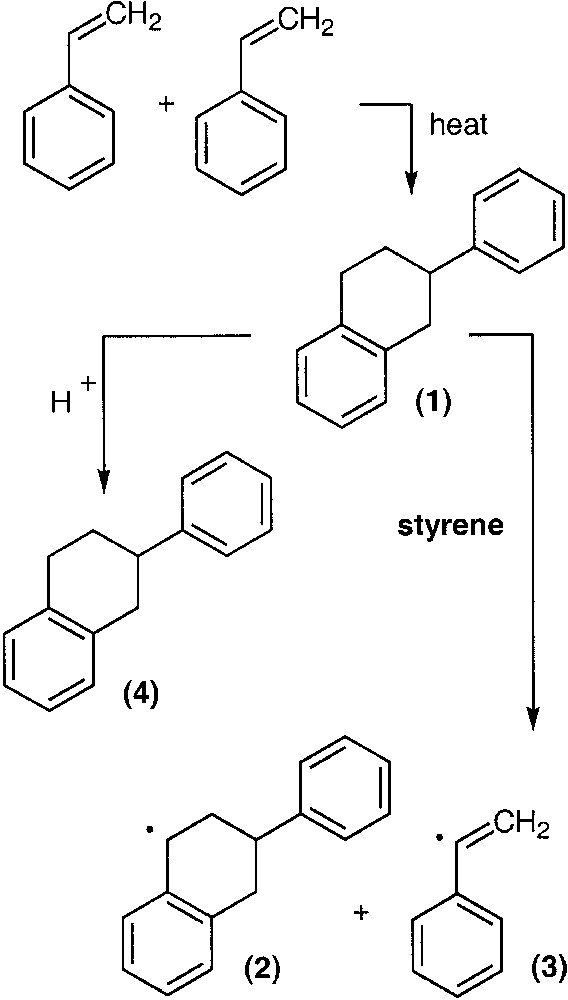
Mayo mechanism of thermal styrene polymerization.
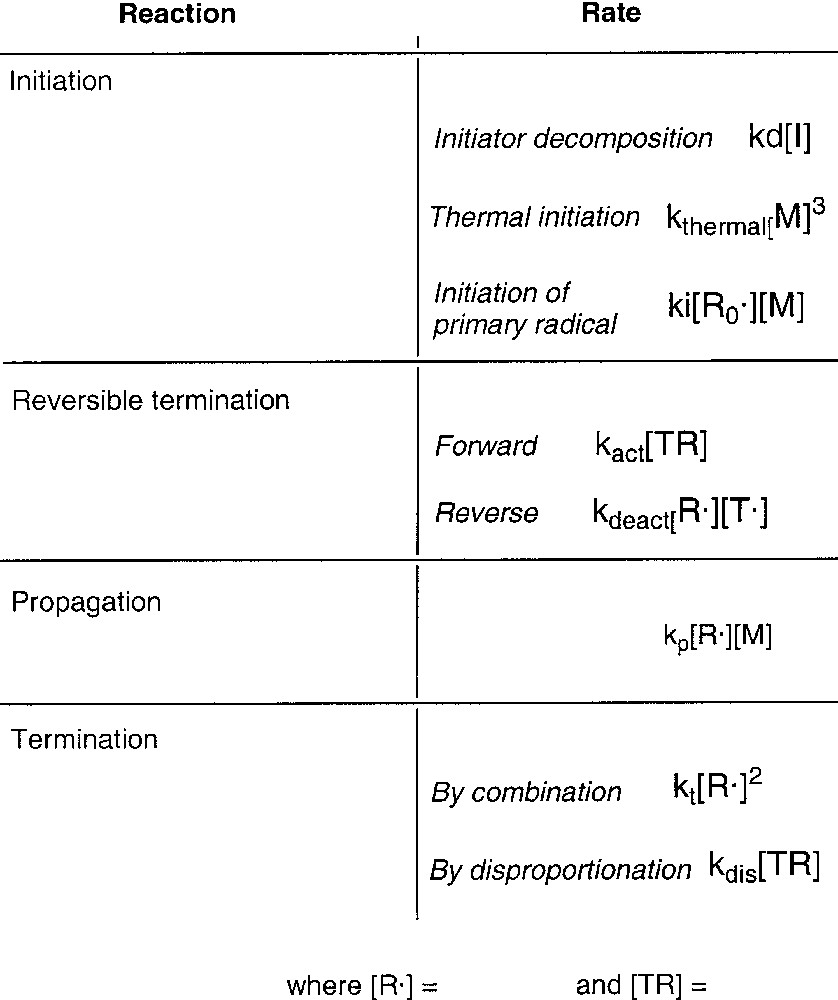
Primary reactions in nitroxide-mediated radical polymerization.
3 Emulsion and miniemulsion polymerization
Ideally NMRP could be performed in emulsion, which is a simpler and more commercially established process than miniemulsion polymerization, albeit less versatile. In traditional emulsion polymerization, an aqueous phase consisting of water, surfactant and buffer is mixed with an organic phase consisting of ‘water-insoluble' monomer (the monomer may have low to moderate solubility in the aqueous phase, but readily disperses as a distinct phase) and perhaps chain transfer agents or cross-linking agents. Initiator is usually soluble in the aqueous phase, although it is possible to use monomer-soluble initiators. Most of the monomer disperses in droplets 1–20 μm in diameter, while a small portion swells the surfactant micelles. Upon heating the mixture, initiator decomposes yielding primary radicals in the aqueous phase. In micellar nucleation, monomer adds to these radicals until they reach a critical length to become sufficiently hydrophobic to enter a micelle, thereby nucleating a particle. Homogeneous nucleation can occur when the surface area of the micelles and particles is low; propagating aqueous oligoradicals may add enough units to precipitate from solution before being ‘captured' by micelles or particles, thus forming the precursor for a polymer particle. Emulsion polymerizations are usually run with surfactant concentrations above the critical micelle concentration so that micellar nucleation dominates. As the monomer in the new particles is consumed, the droplets act as monomer reservoirs; monomer diffuses from droplets through the aqueous phase to the particles, maintaining near constant monomer concentration in the particles until the droplets are consumed. In the final stages, the remaining monomer in the particles is polymerized. Emulsion polymerization readily allows near-complete conversion (>99%) to be achieved in short reaction times. However the nucleation step is sensitive to several factors, and variability in the number of particles nucleated translates into variation in the final particle size distribution. Furthermore because the reaction rate increases with particle number, variability in the rate and heat removal requirements are also a concern.
Miniemulsion polymerization, pioneered by El-Aasser's research group at Lehigh University [7] employs a costabilizer and high shear mixing to reduce the size of the monomer droplets to 0.05–0.5 μm. The droplets are nucleated, and ideally a 1:1 mapping is obtained between the initial monomer droplet size distribution and the final polymer particle size distribution. In practice, not all of the droplets may be nucleated and some homogeneous nucleation can occur, depending on choice of reaction conditions. (Surfactant concentrations are maintained below the critical micelle concentration to eliminate micellar nucleation.) The costabilizer can be either a hydrophobe (e.g., long-chain alkane), which thermodynamically reduces the tendency to Ostwald ripening (diffusional degradation) of small droplets with high surface energies, or it may be a long chain alcohol (e.g., cetyl alcohol) that partitions at the particle interface, reducing interfacial tension and providing a barrier to droplet coalescence, as well as acting to reduce Ostwald ripening. Because the particle size distribution is not determined by a sensitive nucleation step as with emulsions, reproducibility of the particle size distribution and therefore reaction rate is excellent. A variety of monomer-soluble additives (liquid and solid) can be easily incorporated into particles using miniemulsion polymerization, while this is not possible in emulsion because of the necessity of transporting the additive through the aqueous phase from droplets to particles. Miniemulsions also allow the polymerization of highly water-insoluble monomers that are difficult to polymerize in emulsion because of their slow transport rates through the aqueous phase. There are two features of miniemulsion polymerization that may disadvantage it in comparison to emulsion: use of a volatile costabilizer that is undesirable in the final product, and the necessity of using a high-shear mixing step. However, Schork has shown that polymer can function adequately as a costabilizer, eliminating the first concern [8]. There are now available (at industrial scale) in-line high-shear mixers capable of providing the required shear rates to make submicron droplets, thus reducing the concern about an additional process step.
4 Nitroxide-mediated polymerizations in emulsion
Initial efforts at conducting NMRP in heterogeneous systems attempted to use emulsion polymerization [9–12]. However, severe problems with colloidal stability were invariably experienced, ranging from complete coagulation of the latex at low conversions to the presence of large (>1 μm) particles. Some success was achieved using 4-amino-2,2,6,6-tetramethyl-1-piperidinyloxy (69% conversion in 36 h, Mn = 6.0 k, polydispersity = 1.7) and an amino alkoxyamine (69% conversion in 36 h, Mn = 42 k, polydispersity = 1.7) [10]. These polydispersities are higher than typical values for bulk SFRP polymerizations (1.1–1.3). Using four differently substituted TEMPO derivatives of varying water solubilities, Cao et al. [12] were able to achieve reasonably well-controlled reactions in styrene emulsion polymerization. Significant differences were observed for the different nitroxides. Very low water solubility of the nitroxide resulted in an uncontrolled polymerization, while too high water solubility resulted in slow aqueous phase initiation that hindered the polymerization rate. Using ATEMPO (4-acetoxy-2,2,6,6-tetramethyl-1-piperidinyloxy), 81% conversion was reached in 12 h, giving a polymer with Mn = 18k and polydispersity ~1.3. A small portion (~2–10%) of high molecular weight polymer was also observed. The proportion of the high molecular weight was higher at low conversions, and was probably caused by some particles being nucleated without nitroxide. The latex was reported to be stable, with particle sizes < 100 nm. However it is important to note that complete particle size distributions were not reported, and therefore it is premature to conclude that emulsion polymerization was successfully conducted, since it is colloidal instability that provides the greatest challenge in these systems.
The reasons for the colloidal stability problems in NMRP emulsion polymerization have often been attributed to the nucleation step and/or the presence of polymerization in the monomer droplets. Unquestionably some of the problems have been related to difficulties in stabilizing very small particles in the early stages of polymerization. This may be compounded by the unusual condition in NMRP systems that in the early stages of reaction there is a high concentration of polymer chains (all chains are initiated at the outset of reaction), and they are oligomeric. In contrast, with conventional emulsion polymerization the chain concentration is much lower and most are high molecular weight. This problem is related to the effectiveness of the chosen surfactant system, and can probably be resolved with selection of appropriate surfactant(s).
Of much greater concern is the thermal polymerization that occurs in the monomer droplets. In itself this is not overly serious, since droplet polymerization also occurs in conventional emulsion polymerization. The real issue is lack of compartmentalized kinetics in heterogeneous NMRP systems coupled with droplet polymerization. In conventional emulsion polymerization, the polymerization rate in the particles in much higher than in the droplets because the growing chains are segregated from each other. Therefore the droplets will always have a lower polymer concentration than particles and will continue to act as reservoirs. (Conventional styrene autopolymerization in emulsion yields stable latexes.) However with NMRP, compartmentalization effects are not observed [13–16] and therefore the polymerization rate and polymer concentration in the droplets and particles are the same. Consequently there is no thermodynamic driving force for monomer diffusion from droplets to particles, and so the monomer droplets do not disappear. Unpublished results from our laboratory have demonstrated we can conduct NMRP emulsion polymerizations up to ~60–70% conversion to obtain a latex consisting of a population of 50–100 nm particles, but also a population of ~1–5-μm particles. These larger particles tend to settle out slowly, forming a sticky layer of polymer on the reactor internals. If we continue the polymerization, more of the small latex particles (which incidentally are quite stable) coalesce with the larger particles until complete coagulation occurs. Our results, consistent with the findings of other authors, showed that the living radical polymerization continues in a well-behaved manner even after colloidal instabilities occur. We have also conducted seeded styrene NMRP emulsion polymerizations beginning with different initial amounts of monomer (non-living seed polymer). If droplets are initially present, the existence of large particles and coagulation persist as problems. However if no monomer droplets are initially present, the polymerization can be conducted with the preservation of the original particle size distribution.
At present, a successful NMRP emulsion polymerization system has not yet been demonstrated. To be considered successful, preservation of both the living nature of the polymerization and elimination of oversize particles needs to be established. Creative solutions may be found that could enable NMRP emulsion polymerization, including running the polymerization in a starved mode so that no monomer droplets exist. However, the added complexities in conducting the process may exceed the slight additional complexity in using miniemulsions, which have demonstrated far greater robustness for NMRP.
5 Nitroxide-mediated polymerizations in miniemulsion
5.1 Initiation by bicomponent systems
Using a bicomponent initiating system, consisting of a nitroxide and a free-radical initiator such as benzoyl peroxide (BPO) or potassium persulfate (KPS), is a simple and reasonably effective approach to conducting nitroxide-mediated polymerizations. Upon thermal decomposition of the initiator to yield two primary radicals in the first few minutes of reaction, an alkoxyamine is formed in situ by the addition of one or more monomer units and subsequent deactivation by nitroxide. In-situ formation of the alkoxyamine eliminates the need to synthesize and purify alkoxyamine, which can be a time-consuming and sometimes difficult step. However, there can be considerable variation in the number of chains initiated as reaction conditions are changed in bicomponent systems [3,16–19]. The nitroxide-to-initiator ratio is critical to the progress of the polymerization, affecting both the kinetics and molecular weight. Too much nitroxide causes long induction periods due to the excess nitroxide present at the outset of reaction, while uncontrolled polymerization results if the nitroxide:initiator ratio is too low. Nitroxide:initiator ratios of ~1.1–1.8 have been found to give an optimal balance, providing reasonably fast rates while maintaining a controlled polymerization. Achieving effective control of the molecular weight using bicomponent systems is more difficult with heterogeneous systems than homogeneous systems because of the different nature of the chain initiation mechanism. Chain initiation in bulk or solution involves only one phase, and is therefore relatively predictable and not as sensitive to factors such as changes in the initiator or nitroxide type. However in heterogeneous systems, the initiation mechanism can be more complex.
In a miniemulsion, nitroxide and initiator partition between the aqueous and organic phases. The partition coefficients for TEMPO, 4-hydroxy-TEMPO and 4-amino-TEMPO have been reported [20], but such data is often not available. Either monomer-soluble or water-soluble initiators may be used in miniemulsion NMRP, although the locus of initiation is likely different. Using monomer-soluble initiators such as BPO, the chains will be predominantly initiated in the monomer droplets. The limited water solubility of BPO may lead to a small fraction of chains being initiated in the aqueous phase. When a water-soluble initiator such as KPS is used, almost all chains will originate in the aqueous phase, except for those derived from styrene thermal initiation. After addition of at least one monomer unit to a primary radical in the aqueous phase, the oligoradical may be deactivated by nitroxide, propagate or terminate with another radical. After reaching a critical length (probably 2–3 monomer units in the case of styrene), an active oligoradical or alkoxyamine (dormant chain) may enter a droplet/particle. If a radical enters, it is likely to propagate, thus becoming too hydrophobic to subsequently exit the particle. If an alkoxyamine enters a particle however, its absorption may be reversible since it will take on average ~102–103 seconds to dissociate and then add monomer. The hydrophilicity of the nitroxide and the nature of the initiator end-group will be important in determining the critical length for entry of an oligoradical or alkoxyamine into a droplet/particle. It has been shown that when the relatively water-soluble 4-hydroxy-TEMPO is used to mediate styrene miniemulsion polymerization, the observed number of chains (as determined from molecular weight measurements made by gel permeation chromatography) continued to increase for a prolonged time regardless of whether KPS or BPO was used as initiator [18,19]. However when TEMPO was used, the number of chains was nearly constant from the outset of polymerization (Fig. 1 ). This behavior is consistent with slow entry of 4-hydroxy-TEMPO-terminated aqueous oligoradicals into particles. Because of the greater solubility of the 4-hydroxy-TEMPO, longer times were required to reach the critical length for entry, while with the more hydrophobic TEMPO, critical entry lengths were reached quickly.
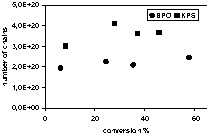
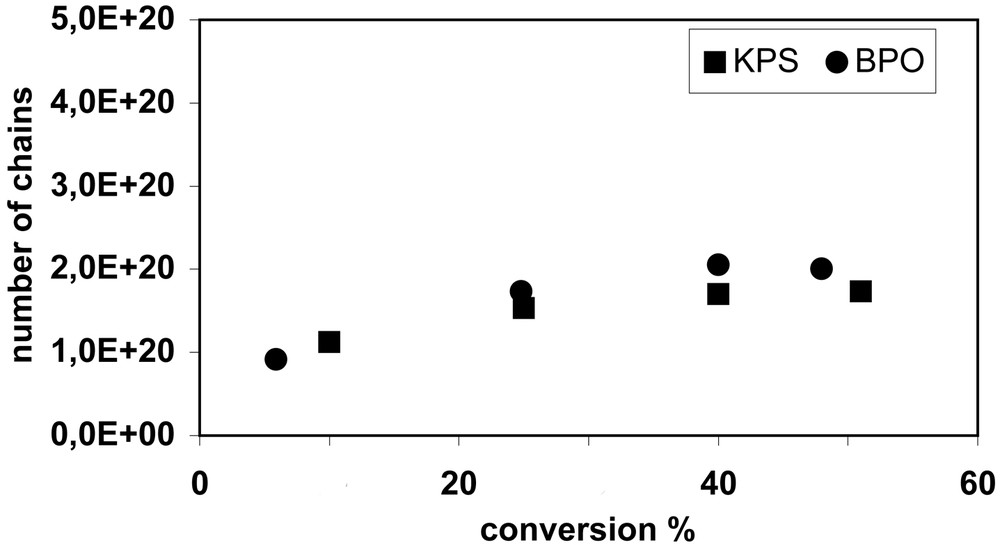
a. Variation of the total number of chains as a function of conversion for TEMPO-mediated runs (0.4 mmol initiator, TEMPO/initiator = 1.7). b. Variation of the total number of chains as a function of conversion for OH-TEMPO-mediated runs (0.4 mmol initiator, OH-TEMPO/initiator = 1.7).
Tortosa et al. conducted a systematic study of the effects of varying the water solubility of both nitroxide and initiator [19]. Polymerizations were initiated with KPS or BPO in the presence of either TEMPO or 4-hydroxy-TEMPO. Four different bicomponent initiating systems were therefore studied (BPO/TEMPO, BPO/4-hydroxy-TEMPO, KPS/TEMPO, KPS/4-hydroxy-TEMPO). Miniemulsion styrene polymerizations were undertaken by separately varying the initiator concentration and the nitroxide:initiator ratio in each of the four systems. Initiator efficiency, which determines the number of polymer chains and hence molecular weight, and the final conversion were found to be strongly affected by both the amount and type of nitroxide in the system, as well as the choice of water-soluble versus oil-soluble initiator. Selection of appropriate conditions allowed synthesis of polystyrene with low polydispersity (PDI < 1.25) and number average molecular weight up to 50 000 g mol–1 in 6 h reaction time.
The respective partition coefficients between styrene and water are 2.2 M/M for 4-hydroxy-TEMPO) and 98.8 M/M for TEMPO [20], where M/M is moles of nitroxide in styrene per moles of nitroxide in water. In Tortosa's experiments, approximately 98% of the unbound TEMPO was calculated to be in the droplet/particle phase, while only ~36% of the unbound 4-hydroxy-TEMPO resided in the droplet/particle phase. At a nitroxide:initiator ratio of 1.7:1, the conversion plots, –ln(1 – x) versus time, were approximately linear with the non-zero intercept indicating the presence of an induction period of ~1 h (Fig. 2 ). The linearity indicates the expected living behavior. In the case of polymerizations mediated by TEMPO, nearly identical conversion versus time behavior was observed using either BPO or KPS. With 4-hydroxy-TEMPO, the kinetic plots were also very similar for the BPO- and KPS-initiated runs. All four bicomponent systems show nearly identical kinetics, which can be attributed to thermal polymerization playing a major role in determining the overall polymerization rate, as has been previously observed in nitroxide-mediated styrene polymerizations [21,22]. Charleux also observed induction periods of ~45 min in styrene miniemulsion polymerizations initiated by a redox system (Na2S2O5/K2S2O8) and mediated by SG1 (nitroxide:initiator = 1.2) [3]. The induction period was attributed to free SG1, and it was also noted that aqueous phase propagation would be slow, resulting in the slow growth and transfer of aqueous alkoxyamines to the particle phase. It was unclear whether or not this slow entry into the particles would affect the rate. However, in Tortosa's experiments, there was not a significant difference observed in the length of the induction periods when BPO was used instead of KPS. Because the BPO-derived radicals would be mostly in the particles, high initial levels of excess nitroxide resulting from inefficient initiation are probably primarily responsible for the induction periods and not slow growth of oligoradicals in the aqueous phase. Furthermore, it is curious that no significant rate differences were observed in the length of the induction periods between the TEMPO- and 4-hydroxy-TEMPO-mediated runs, despite their widely varying partition coefficients. This is consistent with the findings from our calculations of nitroxide diffusion rates that showed for emulsion/miniemulsion size particles, nitroxide diffusion between phases would be sufficiently fast to maintain equilibrium [23]. With 4-hydroxy-TEMPO, it is clear that entry into the particles was much slower than with TEMPO, as described above, which does affect the molecular weight profile. However the kinetics are not affected.
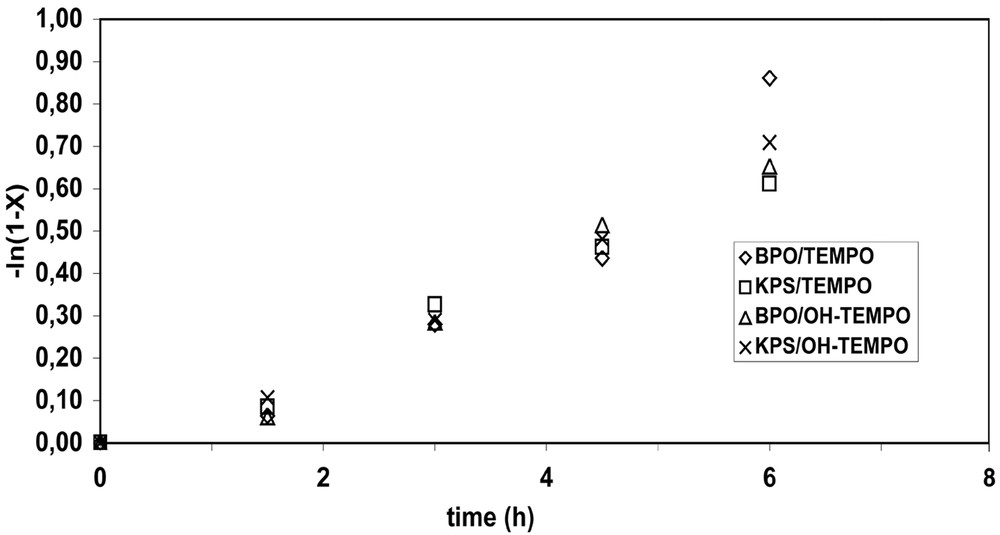
–ln(1 – x) versus time for BPO- or KPS-initiated runs mediated by either TEMPO or OH-TEMPO (0.4 mmol initiator; nitroxide:initiator = 1.7).
While the rate is insensitive to the choice of initiator for these reaction conditions, the number of chains initiated and the Mn vs. conversion profiles are strongly affected by the choice of both initiator and nitroxide. For TEMPO-mediated runs, the Mn plots are linear with conversion and have an intercept near zero. Mn is higher at a given conversion for the BPO-initiated runs than for the KPS-initiated runs, indicating higher initiation efficiency for KPS (Fig. 3 ). For 4-hydroxy-TEMPO-mediated runs, Mn versus conversion profiles are nearly independent of initiator type, indicating the same number of chains is initiated with either BPO or KPS. Furthermore the plots of Mn versus conversion display a non-zero intercept, although they are linear, suggesting the polymerization was poorly controlled in the early stages, probably due to low 4-hydroxy-TEMPO concentration in the particles. For either TEMPO or 4-hydroxy-TEMPO mediated runs, the final polydispersity varied in the range from 1.08 to 1.26.
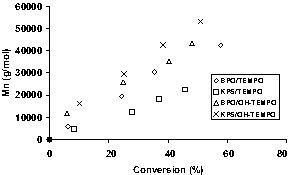
Mn versus conversion for BPO- or KPS-initiated runs mediated by either TEMPO or OH-TEMPO (0.4 mmol initiator; nitroxide:initiator = 1.7).
For each of the four bicomponent initiating systems, polymerizations were also run at two initiator levels (0.4 or 1.24 mmol) and two nitroxide:initiator ratios (1.7 or 3.6). The low nitroxide:initiator ratio (1.7) represents a slight deficiency of nitroxide molecules compared to the number of radicals arising from the decomposition of the initiator, and the high ratio (3.6) corresponds to an excess of nitroxide. For BPO-initiated polymerizations, there were not significant differences in kinetic behavior between the two nitroxides, while KPS-initiated polymerizations were more influenced by the water solubility of the nitroxide. Induction periods can be seen in most of the –ln(1 – x) versus time plots, indicating the presence of excess nitroxide at the beginning of the polymerization. Differences in the lengths of the induction periods are due to different amounts of excess nitroxide present after initiator decomposition. When the nitroxide:initiator ratio is high (3.6), longer induction periods (> 2 h) are observed. Fig. 4 shows plots of conversion for the KPS/4-hydroxy-TEMPO system, illustrating the large differences in conversion at different [KPS] and 4-hydroxy-TEMPO:KPS ratios, while Fig. 5 illustrates that differences in Mn are considerably smaller, and reflects changes in initiator efficiency.
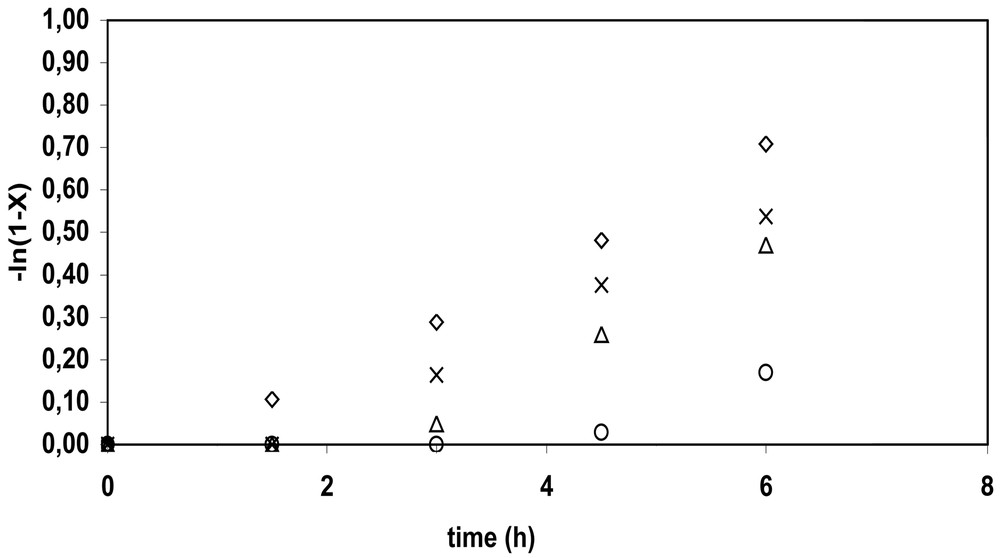
Effect of variation of KPS and OH-TEMPO concentrations on conversion (◊: 0.41 mmol KPS, OH-TEMPO:KPS = 1.7, ×: 1.24 mmol KPS, OH-TEMPO:KPS = 1.7, △: 0.41 mmol KPS, OH-TEMPO:KPS = 3.6, ○: 1.24 mmol KPS, OH-TEMPO:KPS = 3.6).
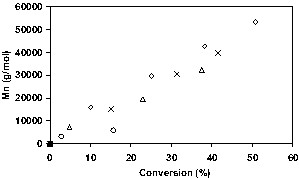
Effect of variation of KPS and OH-TEMPO concentrations on molecular weight. Legend as in Fig. 4.
Tortosa found that initiator efficiency varied considerably between the four bicomponent initiating systems, and also showed significant variation within a given system as conditions were varied. Initiator efficiency is defined as:
| (1) |
The range of F is considerable, from ~0.1–1, depending on the experimental conditions (Fig. 6 ). For the TEMPO-mediated polymerizations at a given set of conditions, F is higher for KPS-initiated runs than BPO-initiated runs. In contrast, with the 4-hydroxy-TEMPO-mediated runs there are not significant differences between the two initiators. TEMPO-mediated runs usually have higher F than runs mediated by 4-hydroxy-TEMPO. For a given bicomponent system, F may vary by a factor of > 2, as the initiator concentration and nitroxide:initiator ratio are varied. Higher initiator concentrations result in lower efficiencies, while higher nitroxide:initiator ratios raise efficiencies, although at the cost of longer induction periods and lower final conversions.
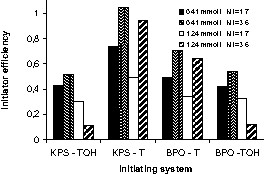
Initiator efficiency at t = 6 h for the bicomponent initiating systems at different initiator concentrations and nitroxide:initiator ratios.
While reasonably good results have been obtained with bicomponent systems in miniemulsion NMRP, the sensitivity of initiation efficiency to reaction conditions and initiator/nitroxide selection makes it difficult to obtain predictable molecular weights. Despite their attractive simplicity, if controlling molecular weight is critical, alkoxyamine-initiated systems are more desirable.
5.2 Rate enhancement
The low polymerization rates of NMRP reactions compared to conventional free radical polymerizations prompted the use of rate-enhancing additives, including organic acids such as camphorsulfonic acid (CSA), to accelerate bulk NMRP polymerizations [24–27]. The addition of CSA to TEMPO-mediated styrene polymerizations yields higher molecular weight in less time, but at the expense of a broader molecular weight distribution if the CSA concentration is too high. The role of CSA is not straightforward, and it is known to have three separate effects. The most important is direct consumption of free nitroxide, as shown by Veregin et al. using in situ ESR measurements in bulk styrene NMRP [24]. Nitroxide disproportionation by the acid was believed to be the most probable route for TEMPO consumption. Lower nitroxide concentration shifts the equilibrium in favor of active radicals, thereby increasing the polymerization rate. Veregin et al. also found the rate constant for the deactivation between a growing radical and nitroxide is decreased in the presence of acid [24] based on their observation of a ~30% increase in the ratio of the propagation rate parameter to the equilibrium constant for deactivation (kp/K) in the presence of CSA. Finally, the thermal polymerization of styrene is influenced by CSA and other organic acids. Buzanowski et al. demonstrated that, in the presence of acid, the dimer 1 in Scheme 2 is reduced to 4 [28]. Georges et al. studied the autopolymerization of styrene in presence of TEMPO with and without added CSA [25,29]. Less low molecular polymer was seen when CSA were used, which was taken as evidence of reduced thermal initiation. The number of thermally initiated chains was reportedly reduced by a factor of 4 with CSA.
CSA has been used in bicomponent NMRP miniemulsions by Prodpran et al. [17] and Cunningham et al. [30]. Cunningham et al. conducted an extensive study of the effects of CSA in BPO-initiated styrene NMRP mediated by TEMPO or 4-hydroxy-TEMPO under a range of conditions. The CSA:nitroxide ratios used in their study ranged from 0.5 to 5.0. The observed effects of CSA addition on conversion and molecular weight did not generally show significant differences between TEMPO and 4-hydroxy-TEMPO mediated polymerizations, despite the vastly greater tendency of 4-hydroxy-TEMPO to partition into the aqueous phase. The dominant role of thermal initiation in determining the radical concentration is likely why few differences are observed between the two nitroxides.
CSA addition moderately increased conversion at nitroxide:BPO = 1.7 but had a much more pronounced effect at nitroxide:BPO = 3.6. The larger effect is expected when the nitroxide concentration is initially high, as there is more nitroxide available to consume. The effects of the CSA concentration on conversion and molecular weight are shown in Fig. 7 for TEMPO-mediated polymerizations (TEMPO:BPO = 2.7). Early in the polymerization, conversions are higher with increasing [CSA]. However as the polymerization progresses the differences become much less pronounced. The effects of CSA on molecular weight at a given conversion are minimal for CSA:nitroxide ratios of 0.5 or 1.0 but become more significant at ratios of 1.5 and 5.0. As more CSA is added, fewer chains are initiated because of nitroxide consumption. Conversion is enhanced without any significant increase in polydispersity except at the highest concentration of CSA. Interestingly, CSA addition usually resulted in a decrease in particle diameter, i.e. more miniemulsion droplets were nucleated in the presence of CSA. The cause is unknown but may be attributable to lower nitroxide concentrations in droplets that allows thermal initiation to proceed more readily in monomer droplets, thereby leading to increased nucleation.
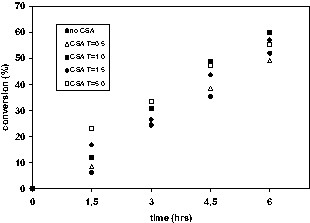
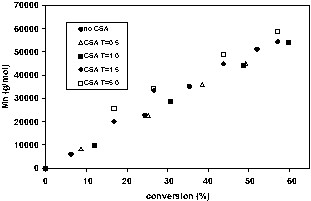
a. Conversion versus time in the presence of varying CSA:nitroxide ratios for TEMPO-mediated polymerizations (TEMPO:BPO = 2.7). b. Number average molecular weight versus conversion in the presence of varying CSA:nitroxide ratios for TEMPO-mediated polymerizations (TEMPO:BPO = 2.7).
CSA can be expected to partition to some extent into the aqueous phase during polymerization, reducing the concentration inside the particles. Prodpran et al. reported CSA being less effective in rate enhancement in miniemulsion polymerizations than in bulk [17]. Studies in bulk systems reported in the literature report significant rate enhancements with CSA:nitroxide ratios of~ 0.5–1.0, but the magnitude of the rate enhancement in miniemulsion is smaller. For example, Veregin et al. observed an increase in the six hour conversion from 50% to 80% in bulk using CSA:TEMPO = 0.34 (TEMPO:BPO = 1.3) [24]. This represents a much greater increase then observed in our experiments even though we used higher CSA:TEMPO ratios, and a TEMPO:BPO ratio of 1.7, which should give a larger effect than a TEMPO:BPO ratio of 1.3. These results are consistent with reduced CSA concentrations in the particles due to phase partitioning, indicating higher CSA:TEMPO ratios concentrations are required in miniemulsion to achieve the same degree of rate enhancement seen in bulk.
5.3 Initiation by unimer (alkoxyamine) systems
The use of bicomponent initiating systems creates the challenge of effective molecular weight control because of the variable nature of the initiation efficiency. Recent work has explored the use of unimers in miniemulsion NMRP [15,31–33]. Pan et al. [15,31] used a TEMPO-terminated oligomer of polystyrene (TTOPS) as a polymeric initiator that has the advantage of not having even marginal solubility in the aqueous phase (free nitroxide can of course partition into the aqueous phase). In their first study, styrene was polymerized at 125 °C using a TTOPS initiator with Mn = 7050 at loadings of 5 wt% and 20 wt% in the organic phase. A linear relationship was seen between Mn and conversion up to the final conversion (~75%). Experimental molecular weights were consistently lower than theoretically expected values, with the discrepancy increasing as time progressed. This may have been caused by an increasing chain number due to thermal initiation. The final polydispersities seem relatively high for NMRP, at 1.76 for the 5% TTOPS run (76% conversion), and 1.86 for the 20% TTOPS run (77% conversion), which may be due to the presence of dead chains in the synthesis of the TTOPS. Throughout the run, lower polydispersity was observed for the higher TTOPS concentration, which is consistent with thermal initiation having less proportional impact because of the higher number of chains.
The role of surfactant (DOWFAX 8390; disulfonated alkyl diphenyl oxide sodium salt) concentration was subsequently studied by Pan et al. [31] using the same system described above, except with a lower molecular TTOPS (Mn = 1500). Final polydispersities were lower than in the study above (~1.4), and theoretical Mn values were close to experimentally observed values. The lower molecular weight TTOPS likely had far fewer dead chains than the TTOPS used previously, which may account for these improvements. Increasing the surfactant concentration from 1.25 to 15 mM resulted in a decrease in mean particle diameter from ~150 to ~60 nm. Particle size was observed to increase with conversion, with more pronounced increases at lower surfactant concentrations. The particle number almost doubled during the course of polymerization at 15 mM DOWFAX, but showed only a very small increase at 1.25 and 2.5 mM. At the lower DOWFAX concentrations, no micelles were present and therefore the small increase in particle number was attributed to homogeneous nucleation. The final particle number was more than an order of magnitude higher at 15 mM DOWFAX compared to 1.25 mM. Despite the large span of diameters and particle numbers, only minor differences were observed in the rate and molecular weight profiles. Rate and molecular weight in conventional emulsion/miniemulsion polymerizations exhibit strong dependence on particle size; the implications of these findings will be discussed further in Section 5.6 on compartmentalization.
A modified, hexadecane-free miniemulsion process was reported by Keoshkerian et al. [32]. They first conducted a TEMPO-mediated bulk styrene polymerization to ~5% conversion to generate a macroinitiator with Mn ~ 2000, and then directly dispersed the partially polymerized bulk mixture into an aqueous phase containing sodium dodecyl benzene sulfonate (SDBS). Although no hexadecane was added, a stable latex (mean diameter ~200 nm) was formed, with the TTOPS acting as both initiator and costabilizer. The conversion was reported to be > 99% in six hours at 135 °C, with a final Mn = 18,789 and PD = 1.15, considerably faster than any previously reported NMRP. A subsequent chain extension with butyl acrylate (BA) to yield block copolymer gave 99.4% conversion of the BA while maintaining narrow polydispersity.
The reason for the surprisingly high rates was not clearly understood. It was speculated that increased droplet nucleation may have contributed through (1) higher average monomer concentrations at the reaction site, and/or (2) less nitroxide accumulation in each particle because of lower chain numbers per particle. These explanations suggest compartmentalization effects are important, but Pan's data [31] suggests otherwise. Smith et al. [33,34], in studying the same system, noticed that at lower [SDBS] of 0.021 M, there was only moderate rate improvement with the hexadecane-free process. Because the overall polymerization rate is determined largely by thermal initiation, removal of the hexadecane leads to increased rate since it results in slightly higher monomer concentration. (Because the thermal initiation rate has a third order dependence on monomer concentration, the effect of removing hexadecane seems disproportionately large). However at higher [SDBS] of 0.089 M, similar results to Keoshkerian et al.'s ones were obtained, with conversions > 95% obtained in 3 h while maintaining polydispersities ~1.3 (Fig. 8 ). The final particle size distribution was nearly identical at the low and high [SDBS] (mean diameter ~ 150 nm). This observed insensitivity of particle size to [SDBS] is in contrast to Pan's results with DOWFAX but consistent with previous experience in our laboratory.
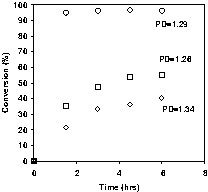
Hexadecane-free miniemulsion polymerizations initiated with TEMPO-terminated polystyrene (○) no hexadecane, 10.6 mM TTOPS (Mn = 19k), 0.089M SDBS (□) no hexadecane, 10.6 mM TTOPS (Mn = 19k), 0.021M SDBS (◊) with hexadecane, 10.6 mM TTOPS (Mn = 19k), 0.021M SDBS.
It remains unclear what causes the dependence of rate on [SDBS]. The effect may be attributable to either the SDBS itself or impurities (e.g., sulfonic acid) that may consume nitroxide. We have determined that different batches of SDBS display very different kinetic behavior when used in miniemulsion NMRP. We have also conducted experiments using DOWFAX 8390. Over a wide range of concentrations (0.021–0.12 M), we found that variation in DOWFAX 8390 concentration did not significantly affect the rate. The DOWFAX-stabilized runs all had lower conversion than any of our SDBS stabilized runs (yielding ~40% conversion in six hours), providing further evidence that the SDBS is influencing the kinetics even when used in low concentrations.
Although the high conversions and short reaction times are encouraging (we have recently obtained >99% conversion as measured by gas chromatography in 2 h with PD ~ 1.3) there needs to be concern about the livingness of the system displaying such high rates. If there is significant loss of livingness, it seems surprising that such low polydispersities are maintained, although polydispersity in itself is not a very sensitive measure of livingness. Chain extension experiments were conducted in which polymer from the final latex was dried, dissolved in styrene, and then further polymerized in bulk. Chain extension experiments are useful but are complicated by the additional loss of chains during the chain extension process. A chain extension experiment with polymer from a miniemulsion polymerization using [SDBS] = 0.089 M revealed that the majority of the chains remain living under these reaction conditions, but there is significant dead chain formation. Work is now in progress in our laboratory to quantify the livingness using a recently developed technique discussed in Section 5.7 [35].
5.4 Colloidal stability
Colloidal stability in NMRP miniemulsions has frequently been a challenge and a concern, particularly for polymerizations run at temperatures >100 °C where traditional surfactants often perform poorly. Sodium dodecyl sulfate, for example, is subject to hydrolysis at these temperatures [10]. SDBS and the structurally similar DOWFAX 8390 are more stable, and have shown good performance as surfactants, giving stable latexes with minimal coagulation. SDBS is available only in a technical grade, and as discussed previously, does influence the kinetics and exhibits batch-to-batch variation in its effects on the kinetics. We heated a non-polymerizing styrene miniemulsion stabilized with SBDS and hexadecane at 135 °C for six hours to determine how stable the droplet size distribution was under these conditions (tert-butylcatechol was added to the styrene to inhibit polymerization) [16]. The mean droplet size increased from ~150 to ~190 nm (Table 1). This experiment was valuable not only in providing a measure of how stable the initial miniemulsion is, but in allowing us to draw conclusions about the fraction of droplets nucleated and/or whether or not additional nucleation occurred.
Stability of styrene miniemulsion monomer droplet sizes under non-polymerizing conditions (T = 135 °C). (Dv = volume average diameter; (D84/D50)0.5 = geometric standard deviation. Measurements made using Malvern Mastersizer 2000
| Time (h) | Dv (nm) | (D84/D50)0.5 |
| 0 | 151 | 1.33 |
| 1 | 173 | 1.42 |
| 2 | 179 | 1.52 |
| 3 | 182 | 1.55 |
| 4 | 190 | 1.61 |
| 5 | 186 | 1.59 |
| 6 | 188 | 1.60 |
In almost all of our experiments with bicomponent initiating systems, the mean particle size was relatively large (>190 nm) with a broad distribution. Given an initial droplet size of ~150 nm, this indicates that in most cases less than half of the droplets were nucleated. However for systems initiated with unimers, the particle sizes are considerably smaller, typically ~120–150 nm, indicating there are more final particles than there were droplets initially [33]. With the comparatively high solubility of styrene in water at 135 °C [20], there is a distinct possibility of homogeneous nucleation. However, the presence of unimer or TTOPS in the droplets at the start of reaction guarantees polymerization will occur in each droplet (several thousand chains are present in each droplet). Examination of El-Aasser's data for bicomponent (BPO/TEMPO) [17] versus TTOPS [15,31] systems at the same surfactant concentration shows the same trend of larger particle sizes with the bicomponent systems. Even when the free radical initiator is monomer-soluble, so that most chain initiation should occur in the droplets and not the aqueous phase, the final particle size is large. This is consistent with our findings that there was no significant difference in final particle size between bicomponent systems using BPO versus KPS, although more nucleated droplets would be expected using BPO [19]. Pan et al. [15] reported an increase in particle number up to ~70% conversion, and then a small decrease suggesting limited aggregation. The effects of varying surfactant concentration on particle size and number were investigated by Pan et al. [31] and discussed in the preceding section. An unusual observation we have made is that low conversion samples (<20%) will often phase separate within a few minutes to give a milky latex with a clear liquid layer on top, presumably monomer. With stainless steel reactors, we cannot see if this liquid layer exists during polymerization. The final latexes did not exhibit this layer unless the final conversion was low. A similar colloidal instability has been reported using RAFT systems in miniemulsion, and a theoretical argument proposed to explain its occurrence [36].
5.5 Modeling and simulation
5.5.1 Interfacial mass transfer
The complexity of conducting living radical polymerizations in miniemulsion, with the existence of phenomena unique to a heterogeneous environment, has posed many questions in the search for explanations to unexpected behavior, particularly where experimental observations differ from bulk NMRP systems or conventional miniemulsion polymerizations. We have conducted a series of mathematical modeling and simulation studies to address critical issues and questions that have arisen in the course of our experimental work [23,37–39]. We had previously measured the partition coefficients of TEMPO, 4-hydroxy-TEMPO and 4-amino-TEMPO at various temperatures, and examined the effect of added polystyrene, hexadecane and SDBS on the partition coefficients to enable us to do quantitative modeling [20].
We began with the basic questions related to diffusion of nitroxide between phases, and the time scales on which this occurs at different times during the polymerization [23]. There are two relevant aspects to this issue. First relates to the time required for diffusion of the nitroxide between phases, allowing for specific characteristics of the system such as particle size and nitroxide diffusivity. The second aspect is how this time compares to the time required for deactivation of radicals by a nitroxide, and thus whether phase equilibrium will be maintained under polymerizing conditions. Specifically, we developed a mathematical model to predict the interfacial mass transfer of nitroxides in a non-reacting NMRP miniemulsion system. Simulations were used to examine how the time required for TEMPO to achieve phase equilibrium is influenced by the TEMPO diffusivity in the aqueous and organic phases, and by average droplet diameter. The analysis was also extended to include 4-amino-TEMPO and 4-hydroxy-TEMPO. Predicted equilibration times were compared with the characteristic time required for the nitroxide to deactivate polymer radicals to ascertain whether the nitroxide will achieve phase equilibrium during polymerization.
5.5.1.1 Influence of TEMPO diffusivity in monomer droplets
The diffusivity of TEMPO in the particles will decrease during polymerization as viscosity increases. If the rate of molecular diffusion is sufficiently reduced, the rate of radical trapping by TEMPO could exceed the rate of mass transfer, resulting in non-equilibrium conditions. Our simulations predict that phase equilibrium is achieved within ~10–4 s. The relatively fast equilibration times can be attributed to the exceptionally large interfacial surface area, which for the system considered in this study, is ca. 8.0 × 103 m2 per liter of miniemulsion. Furthermore, the time required to reach phase equilibrium changes little when the TEMPO diffusivity DTEMPO,drop is varied from 1 × 10–11–1 × 10–8 m2 s–1, but increases significantly when DTEMPO,drop < 1 × 10–11 m2 s–1, indicating that longer equilibration times may be needed at high monomer conversions. The diffusivity of styrene in a solution of 3 wt% styrene/97 wt% polystyrene has been measured to be 3 × 10–11 m2 s–1 at 135 °C [40]. Assuming the diffusivity of TEMPO in polystyrene is comparable to that of styrene, phase equilibrium will still be maintained even at high monomer conversions. Note, however, that in low temperature systems (~90 °C), such as those employing SG1, diffusivity may be significantly lower which may lead to greater equilibration times and possibly non-equilibrium conditions during polymerization.
5.5.1.2 Influence of droplet diameter
Since the rate of interfacial mass transfer is dependent on the total interfacial surface area, process variables affecting average droplet diameter (method of homogenization, surfactant type and concentration, costabilizer) may also influence the rate of interfacial mass transfer. In simulations conducted for a range of average droplet diameters (100–300 nm), we found that phase equilibrium is achieved in < 3 × 10–5 s. However, at larger droplet diameters corresponding to suspension and emulsion polymerizations, the predicted equilibration times are significantly greater (Table 2).
Predicted equilibration times for TEMPO-mediated styrene miniemulsion polymerization systems with varying average droplet diameters (T = 135 °C)
| System | d (nm) | Interfacial surface area (m2 l–1 of system) | Equilibration time (s) |
| Miniemulsion | 200 | 8.0 × 103 | 1.5 × 10–5 |
| Emulsion | 1 × 103 | 1.5 × 103 | 5 × 10–4 |
| Emulsion or Suspension | 1 × 104 | 1.5 × 102 | 5 × 10–2 |
| Suspension | 1 × 106 | 1.5 | 500 |
The relatively slow rates of interfacial mass transfer predicted for emulsion and suspension polymerization systems may lead to conditions where the concentration of TEMPO in either the aqueous or organic droplet phases is insufficient to effectively mediate polymer growth, leading to poorly controlled (non-living) polymerization and increased bimolecular termination. Systems particularly susceptible are those in which the nitroxide and initiator tend to partition preferentially into different phases.
5.5.1.3 Influence of nitroxide partitioning
Since the equilibrium concentration of nitroxide in the aqueous phase and monomer droplets is governed by the partition coefficient of the nitroxide, simulations were performed to investigate the role of the partition coefficients on equilibration times. At 135 °C, the styrene-water partition coefficients of TEMPO, 4-amino-TEMPO and 4-hydroxy-TEMPO are Γj = 98.8, 6.7 and 2.2 M M–1, respectively [20]. Varying Γj was predicted to change the equilibrium concentrations of nitroxide but not the time required to reach phase equilibrium (< 1.5 × 10–5 s).
5.5.1.4 Comparison of equilibration times to deactivation times
If the rate of radical deactivation by TEMPO is significantly faster than the rate of interfacial mass transfer, the nitroxide could deactivate polymer radicals before achieving phase equilibrium, resulting in non-equilibrium concentrations of TEMPO in the aqueous and organic phases. Conversely, if the rate of radical deactivation is slower than interfacial mass transfer, phase equilibrium will be established. The characteristic time required for a nitroxide molecule to react with a polymer radical is:
| (2) |
5.5.2 Modeling alkoxyamine initiated NMRP miniemulsions
A detailed model incorporating NMRP kinetics for alkoxyamine initiated styrene polymerizations was coupled to a mass transfer model to describe miniemulsion NMRP of styrene [37]. The model includes reactions in the aqueous and organic phases, particle nucleation, the entry and exit of oligomeric radicals, and the partitioning of nitroxide and styrene between phases. The influence of nitroxide partitioning on the polymerization kinetics was examined by modeling systems initiated by the unimers BST and OH-BST (BST and OH-BST are benzoylstyryl radicals terminated by the TEMPO and 4-hydroxy-TEMPO respectively as shown in Scheme 4 .). The model was also used to make quantitative estimates of; monomer conversion, number average molecular weight, polydispersity, the molecular weight distributions of active and dormant polymer radicals (derived from both alkoxyamine initiators and thermal initiation), and the molecular weight distributions of living and dead polymer chains.
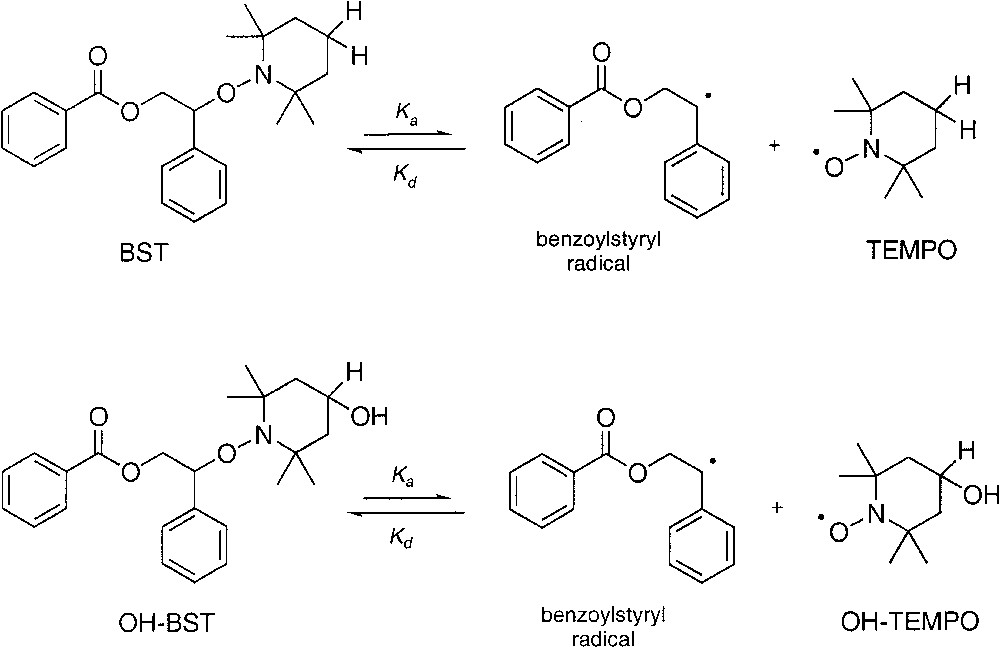
Structure of the two unimers BST and OH-BST [33].
Measured and predicted monomer conversions versus time are shown in Fig. 9 for BST and OH-BST-initiated systems. Conversion is predicted to level off after reaching ~60% due to increasing nitroxide concentration. Monomer conversions are nearly independent of the type of unimer and the initial concentration of BST or OH-BST, a reflection that the rate of polymerization is controlled by the rate of styrene thermal initiation. Experimental and predicted values of Mn are plotted versus conversion in Fig. 10 for BST runs. Mn increases almost linearly with conversion; the slight non-linearity is due to continuous initiation of polymer radicals by styrene thermal initiation, which causes the total number of polymer chains to increase throughout the simulation. This can be seen from the predicted number of polymer chains shown in Table 3.
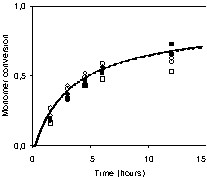
Conversion-time profiles for styrene miniemulsions initiated by BST (open symbols) or OH-BST (filled symbols) (circles, 7 mM; squares, 14 mM; triangles, 20 mM). Lines are model predictions.
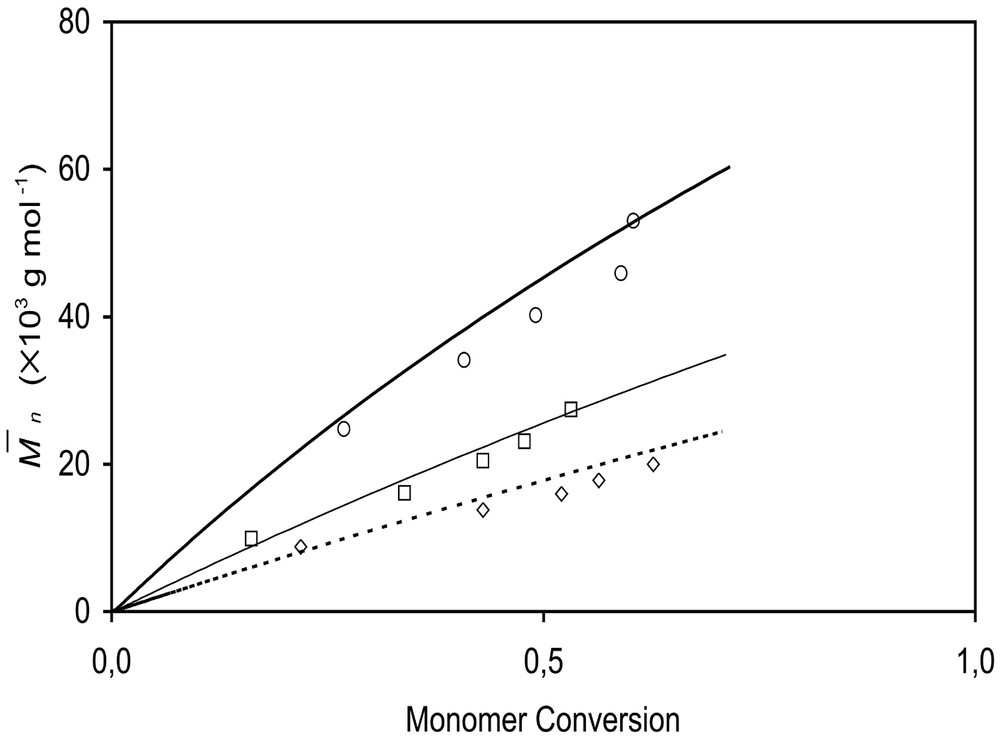
Measured and predicted number average molecular weights, , for styrene miniemulsions initiated by BST. ○, 7 mM; □, 14 mM BST; ◊, 20 mM BST. Lines are model predictions.
Predicted number of polymer chains in BST- and OH-BST-initiated styrene miniemulsion systems at 0%, 20% and 60% monomer conversion
| System | Total number of polymer chains (×1020 chains) | ||
| 0% conversion | 20% conversion | 60% conversion | |
| BST-01 | 1.7 | 1.8 | 2.1 |
| BST-02 | 3.2 | 3.3 | 3.6 |
| BST-03 | 4.7 | 4.8 | 5.1 |
| OH-BST-01 | 1.6 | 1.7 | 2.0 |
| OH-BST-02 | 3.2 | 3.3 | 3.6 |
| OH-BST-03 | 4.6 | 4.7 | 5.0 |
5.5.2.1 Nitroxide partitioning effects
At phase equilibrium, the aqueous phase concentration of nitroxide, [T•]aq, and [T•]org are related through the partition coefficient, Γ = [T•]org /[T•]aq. We had anticipated that [R•]org would be influenced by the value of Γ. 4-hydroxy-TEMPO partitions into the aqueous phase more favorably than TEMPO, which should result in lower values of [T•]org and faster polymerization rates compared to TEMPO-mediated systems. However, the predicted organic phase concentrations of active polymer radicals were found to be nearly equal regardless of whether BST or OH-BST was used to initiate polymerization, which is consistent with the experimental data showing nearly coincidental conversion profiles with BST and OH-BST. Thermal initiation offsets the loss of polymer radicals by termination such that [R•]org exhibits little sensitivity towards Γ. Consequently, the polymerization rate is not significantly influenced by the different partitioning coefficient of the nitroxides. However, in the absence of styrene thermal initiation, both [R•]org and the rate were found to be quite sensitive towards the value of Γ with the rate predicted to be significantly faster in the OH-BST-initiated systems (Fig. 11 ). For monomers that do not thermally initiate as readily as styrene (e.g., butyl acrylate), nitroxide partitioning may greatly influence the kinetics. Recently, Tortosa et al. [41] reported that in the nitroxide-mediated copolymerization of styrene and butyl acrylate in miniemulsion at 135 °C, the rate of butyl acrylate polymerization was found to be significantly faster in systems employing 4-hydroxy-TEMPO compared to systems employing TEMPO. These experimental results corroborate the behavior predicted by our model.
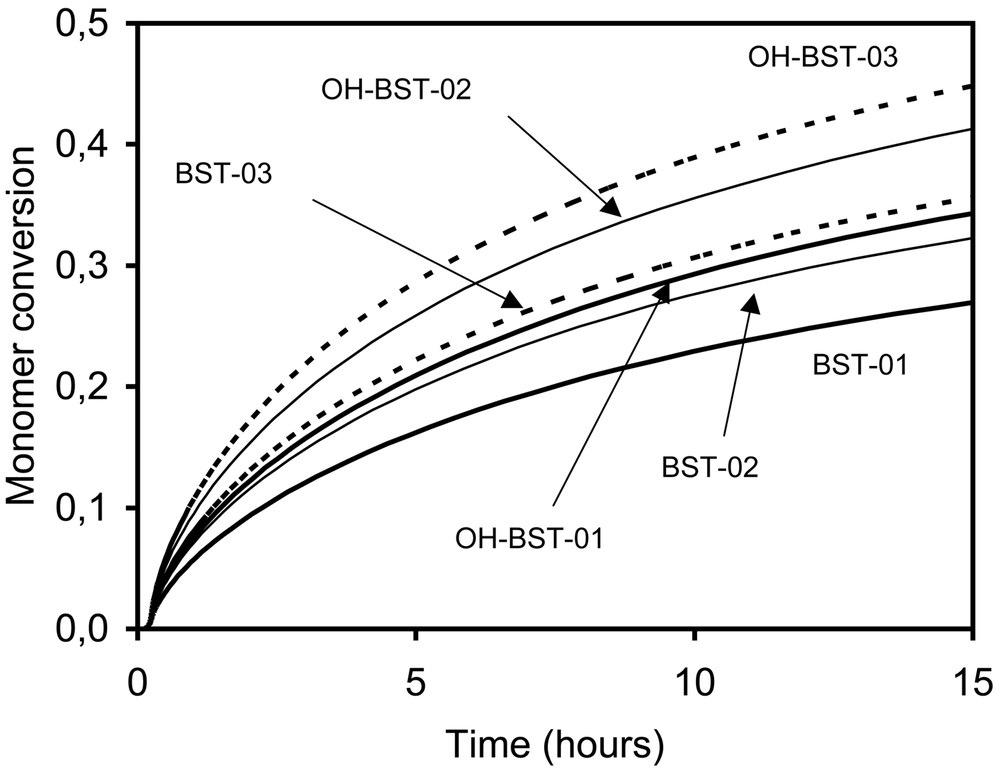
Predicted conversion-time profiles for BST- and OH-BST-initiated styrene miniemulsions in the presence and absence of thermal initiation.
5.5.2.2 Polymer radical distributions and degree of livingness
Our model was designed to keep track of the different radical populations based on how polymer radicals are initiated (unimer homolysis or thermal initiation), how dead polymer chains are created (termination by combination and disproportionation, transfer to monomer, alkoxyamine disproportionation and hydroxylamine disproportionation), and whether living polymer chains are active or dormant. Alkoxyamine disproportionation involves hydrogen abstraction from form the polymer chain end to yield a chain with termination unsaturation and a hydroxylamine, and thus results in the loss of a living chain and a nitroxide [42]. In the presence of trace oxygen, the hydroxylamine can undergo disproportionation to return to a nitroxide. Fig. 12 shows the different polymer chain populations as a fraction of the total number of polymer chains. At the start, dormant IGRs (initiator (unimer) generated radicals) make up 100% of all polymer chains but after 60% monomer conversion, the population of dormant IGRs decreases to only 50% of the total number of polymer chains, with 41% of the polymer chains predicted to be dead. Dormant TGRs (thermally generated radicals) were predicted to make up ~9% of the total polymer chains.
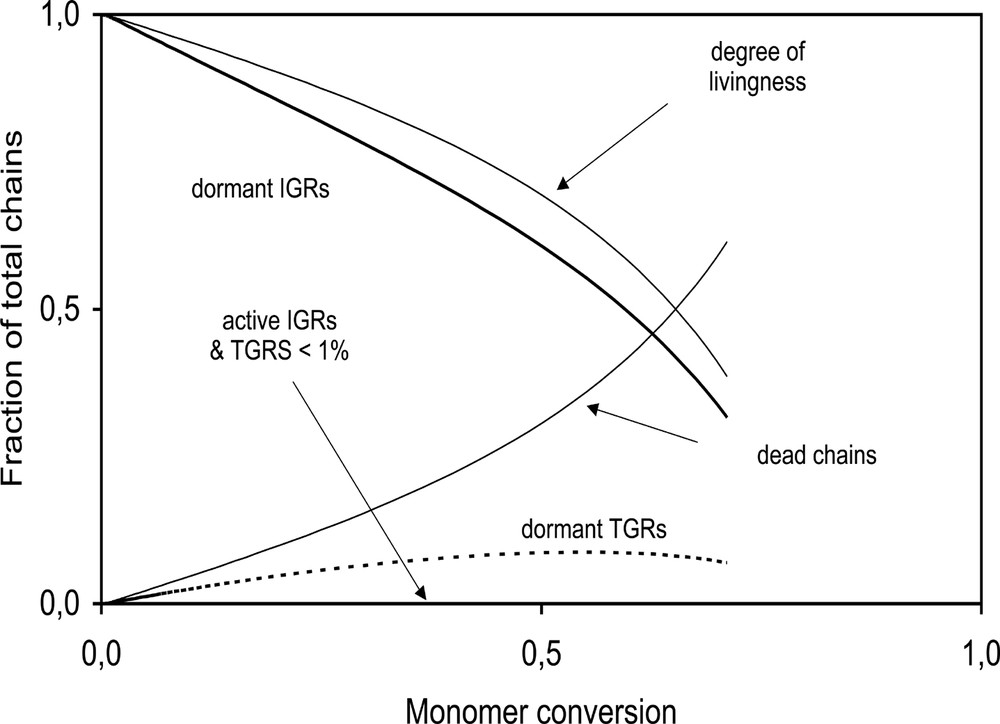
Predicted population profiles of active, dormant and dead polymer chains for system BST-02 (14 mM BST). The predicted degree of livingness (DOL) is also shown.
The continuous decline in the population of dormant IGRs reflects a decay in the overall livingness. We defined the degree of livingness (DOL) as the number of living polymer chains (active and dormant polymer radicals) divided by the total number of polymer chains. Fig. 13 shows that alkoxyamine disproportionation is responsible for terminating the majority of polymer chains and begins to dominate over the other mechanisms early in the simulation. After 60% conversion, ~70% of the dead polymer chains are formed by alkoxyamine disproportionation, while combination and disproportionation, transfer to monomer, and hydroxylamine disproportionation accounted for only 15%, 15% and 1% of the total number of dead polymer chains, respectively.
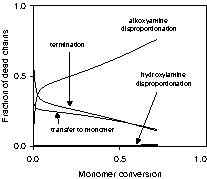
Predicted population profiles of dead polymer chains for system BST-02. Dead polymer chains are formed by termination, transfer to monomer, alkoxyamine disproportionation, and hydroxylamine disproportionation.
The amount of dead polymer chains formed by alkoxyamine disproportionation is disturbingly large. The relatively high reaction temperatures contribute, and lowering the temperature would reduce the disproportionation rate [43]. However this would also extend the reaction time required to achieve a given conversion, thereby increasing the number of dead chains formed by disproportionation. Preserving livingness is complicated by the conflicting constraints posed by minimizing dead chain formation due to irreversible termination versus disproportionation. Minimizing termination requires maintaining a low active radical concentration, which also results in low reaction rates. However the long reaction times required will result in significant dead chain formation via disproportionation. Minimizing dead chain formation due to disproportionation requires reduction of the overall reaction time (higher reaction rates), an objective in direct opposition to that for minimizing biradical termination. An optimized balance must therefore be obtained between losses due to disproportionation and biradical termination.
5.5.2.3 Polymer molecular weight distributions
Fig. 14 shows the number molecular weight distributions (MWDs) of the living and dead polymer chains at 25% conversion. The majority of living polymer radicals are dormant IGRs with a narrow molecular weight distribution. There is a low molecular weight tail due to transfer to monomer. At such a low conversion, the contribution from thermal radicals is small. The MWD of the active TGRs is broad (PD = 1.47) compared to the MWD of the active IGRs (PD = 1.15). The MWDs of the different dead polymer species are broad, with the majority of dead polymer chains formed by alkoxyamine disproportionation, as shown earlier.
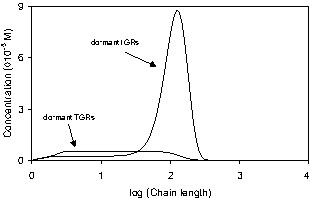
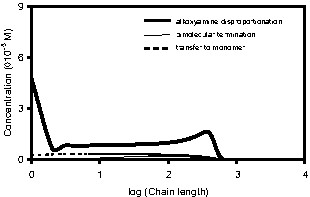
a. Predicted number molecular weight distributions from system BST-02 for living polymer chains at 25% conversion. b. Predicted number molecular weight distributions from system BST-02 for dead polymer chains at 25% conversion.
5.6 Compartmentalization
It has long been recognized that the kinetics and molecular weight obtained in a bulk or suspension polymerization are different than obtained in emulsion polymerization. With particles larger than ~0.5–1 μm, there are enough propagating radicals in each particle to ensure the kinetic equations describing biradical termination are valid. However as particle volume decreases, eventually the number of radicals in each segregated reaction volume (particle) becomes too small (0, 1 or 2 radicals/particle) to be adequately described by the bulk kinetic expressions, and instead balances must be written to account for particles containing a specified number of radicals. The reduction in size changes the primary chain-stopping event from biradical termination to the entry of a radical from the aqueous phase into a particle already containing a radical. This segregation of growing radicals from each other, known as compartmentalization, results in a decrease in the effective termination rate, leading to much higher rates and molecular weights in emulsion/miniemulsion than in bulk. With NMRP, the main chain-stopping event is not biradical termination but rather deactivation by nitroxide. However as particle volume decreases, the number of free nitroxide molecules typically remains high (~101–102). The deactivation step is not affected by reduced particle size and therefore compartmentalization effects are not expected under most conditions.
Butté et al. first discussed compartmentalization in living radical systems, showing that it would operate in reversible transfer systems (e.g., RAFT) but not in reversible termination systems such as NMRP and ATRP [13,44]. They also modeled the NMRP of styrene in miniemulsion, considering both radical and nitroxide compartmentalization [44], predicting that in TEMPO-mediated systems, compartmentalization would lead to fewer dead chains and lower polydispersity but at the expense of a lower rate. The explanation for the lower rate was the loss of the beneficial effects of thermal initiation when the number of free TEMPO molecules per particle became small. For nitroxides with a more favorable equilibrium constant to active radicals, they predicted significant rate enhancement with only a moderate loss of livingness. In a theoretical discussion of living radical polymerizations in dispersed media, Charleux considered the effects of radical compartmentalization for SG1-mediated styrene polymerizations at 90 °C, and concluded that as particle size decreased (below ~100 nm), the rate would increase with a broader molecular weight distribution but fewer dead chains [14]. These conclusions do not apply to cases where thermal initiation is important but are generally applicable for nitroxide-mediated systems for monomers such as butyl acrylate. It is difficult to compare the findings from the above two studies, since they considered different systems, and the role of thermal initiation is probably important.
Pan et al.'s study of surfactant effects in TEMPO-mediated NMRP of styrene in miniemulsion provide a complete set of kinetic and molecular weight data for particles ranging in size from ~150 to ~60 nm [31]. They did not observe significant differences in the conversions or molecular weights, although the smaller particles appear to consistently display slightly higher conversions. This data indicates, as is discussed in their paper, that NMRP systems under their conditions do not exhibit compartmentalization effects, at least down to the lowest particle sizes attained.
5.7 Measurement of livingness
For many applications, maximizing the fraction of living chains is a priority, and yet reliable techniques to quantify livingness are scarce. Chain extension experiments, in which polymer is dissolved in more monomer and then polymerization continued to extend the growing chains, can provide a crude assessment of the degree of livingness by comparing the evolving molecular weight distributions. If a low-molecular-weight shoulder appears, it is evidence of dead chains. However this is not a sensitive technique, and suffers from the loss of more chains during the actual chain extension process. The most common representation of molecular weight distribution data in chain extension tests is the GPC molecular weight distribution (dW(M)/dlogM vs M), where W is the cumulative weight fraction distribution and M is molecular weight. However, the true weight fraction distribution w provides a more discriminating look at chain extension tests. (The two distributions differ by a factor of 1/M; i.e. W(1/M) =w [45]). By emphasizing the higher molecular weight fractions, W reduces the apparent presence of the low molecular weight region where the dead chains reside. Livingness appears better when examining W, but the weight fraction distribution w more clearly highlights the number of dead chains at low molecular weights. Polydispersity is also a relatively insensitive measure of livingness. It should be kept in mind that polydispersities of ~1.1–1.3 are not really ‘narrow' in an absolute sense, and that the absolute span of the molecular weight distribution is considerable.
Scott et al. [35] have recently developed a technique that involves exchange of the nitroxide on the sample chain end with a nitroxide that fluoresces strongly in its alkoxyamine form, but only weakly in its free state (a naphthoyloxy analogue of TEMPO). By measuring the fluorescence of the exchanged sample, the number of moles of living chains can be calculated. Furthermore, coupling of a fluorescence detector with a gel permeation chromatograph led to analyses of the molecular weight distributions of the living chains, while a differential refractive index detector gives the molecular weight distributions of the overall sample (living and dead chains). The population of living chains can thus be directly compared to the total chain population. For a series of styrene miniemulsion polymerizations initiated by the BST [33], the concentration of living chains as a function of conversion is shown in Fig. 15 for three different unimer concentrations. The livingness decreases relatively slowly until ~50% conversion, after which it decreases sharply. However the polymerization rate decreased rapidly after 50% conversion, and the observed sharp decrease in living chain concentration is believed to be primarily a reflection of extensive alkoxyamine disproportionation occurring at long reaction times.
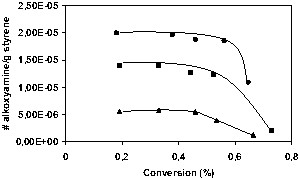
Number of nitroxide-terminated chains as function of conversion for OH-BST initiated styrene miniemulsions [33,35]. (•, 7 mM; ■, 14 mM; ▴, 20 mM OH-BST).
5.8 Acrylate polymerizations
Recent studies have been published on the polymerization of acrylates in miniemulsion [46–48]. With very low rates of thermal initiation compared to styrene, acrylate polymerizations are susceptible to rapid accumulation of nitroxide from termination reactions that suppress the polymerization rate. Therefore successful acrylate polymerization usually requires a means to remove the excess nitroxide, for example by using a nitroxide that spontaneously decomposes slowly (e.g. SG1) or by using an additive to consume the excess nitroxide. Farcet et al. made n-butyl acrylate homopolymers (112 °C and 125 °C) and n-butyl acrylate-co-styrene gradient copolymers (120 °C) in miniemulsion using an SG1-terminated alkoxyamine [46,47]. Excess SG1 was added to some of the polymerizations to provide better control. Conversions of 65–70% were achieved in 6 h when SG1 was added, compared to 92% conversion without added SG1. Final Mn ranged from ~30–50 k. Polydispersities were higher without added nitroxide (1.42) than with SG1 (~1.1–1.4), but these results are still exceptionally good for acrylates. They provided a kinetic analysis that showed, consistent with the experimental findings, that the rate was determined mostly by the initial free SG1 concentration, or more specifically. The copolymers formed were gradient copolymers with a narrow copolymer composition distribution, features caused by the living nature of the polymerization. Because the chains remain alive throughout the polymerization, drift in the monomer composition with time does not lead to composition variation between chains as in conventional radical polymerization, but rather to composition variation along the length of the chain, thus forming gradient copolymers [46,47]. These SG1-terminated polymers also provided a unique tool for differentiating between inter- and intramolecular chain transfer in n-butyl acrylate polymerization [49]. In the n-butyl acrylate homopolymerizations, initiator efficiencies were less than one (~0.8). This unexpected behavior was believed to be caused by a relatively low alkoxyamine activation rate at 112 °C, as well as exit of some of the alkoxyamine initiator into the aqueous phase. Increasing the temperature to 125 °C or using a macroinitiator of Mn = 9550 (prepared using bulk polymerization) were successfully implemented as solutions to each respective problem, and both approaches increased the initiator efficiency to ~1.
Keoshkerian et al. used two different nitroxides in their miniemulsion polymerizations of n-butyl acrylate [48]. They used a polystyrene oligomer (Mn = 1156) terminated with an acyclic nitroxide developed by Hawker [4] as initiator, and added excess TEMPO to give adequate control. Three hours at 135 °C yielded 86% conversion with Mn = 11,816 (polystyrene standards) and PD = 1.27. They also used TEMPO-terminated polystyrene as initiator, with added ascorbic acid, at the same reaction conditions. Three-hour reaction time gave ~60–65% conversion with a final Mn = 12,494 and PD = 1.62. The overall results were not as good using TEMPO but the results were respectable, and the economic attractiveness of using the commercially available TEMPO makes these findings noteworthy, and more generally makes the point that effective control of nitroxide concentration may overcome inherent deficiencies of a given nitroxide.
6 Concluding remarks
Miniemulsion polymerization is a process well suited for nitroxide-mediated polymerizations, yielding stable latexes with the inherent advantages of using aqueous dispersions. With the advent of hexadecane-free NMRP processes, there is little incentive to develop emulsion polymerizations, since additional process steps required to circumvent problems with droplet polymerization would add more complexity than running a miniemulsion. In polymerizations where thermal initiation is important, partitioning effects (especially of the nitroxide) become nearly negligible. However with acrylates and lower temperature styrene polymerizations (< 100 °C), nitroxide partitioning is an important phenomenon. Interfacial mass transfer of the nitroxide is sufficiently fast that phase equilibrium will be maintained, provided particles and/or droplets are < 1 μm. For larger droplets and particles, the surface area may be too small to maintain equilibrium. High conversions (> 99%) narrow polydispersities can now be achieved with styrene miniemulsion NMRP in ~3 h or less, however the livingness of these systems needs to be further investigated.
Acknowledgements
The efforts and contributions of my collaborators Barkev Keosherkian (Xerox Research Centre of Canada), Michael Georges (University of Toronto) and Kim McAuley, graduate students John Ma, Jodi-Anne Smith, Marcus Lin, Catherine Buragina, Hideo Maehata and postdoctoral fellow Karine Tortosa are gratefully acknowledged. Funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Xerox Research Centre of Canada is appreciated.