

1 Introduction
Surface Organometallic Chemistry (SOMC) is an alternative route to functionalize mineral surfaces, such as those of inorganic oxides, zeolites, and supported metals [1–6]... It can have numerous applications in chromatography, molecular separation, new materials for microelectronics, surface treatments for anticorrosion and adhesion purpose, painting technology, glass technology and heterogeneous catalysis.
The first step of SOMC is to synthesize and characterize as precisely as possible ‘relatively’ well-defined surface organometallic complexes. Its second objective is to study the chemical and/or thermal reactivity of these grafted entities. The determination of the stoichiometric reactivity of these well-defined surface organometallic fragments may help us to understand, at a molecular or atomic level, the elementary steps of heterogeneous catalysis [7]. Indeed, it is often difficult to transpose directly the knowledge of molecular chemistry into surface chemistry and so, for obvious reasons, surface organometallic complexes, due to their immobilisation on a rigid surface, may present a chemical and/or thermal behaviour different from their molecular counterparts.
We have shown recently that (n-C4H9)3SnH reacts at room temperature with the hydroxyl groups of a partially dehydroxylated silica with formation of the well defined Si–O–Sn(n-C4H9)3 complex (1) [8], as shown in Eq. (1):
| (1) |
The same reaction also occurs at the external surface of mordenites [5]. The steric hindrance generated by the grafted complex induces a modification of the pore opening of the zeolites and thus leads to a precise regulation, at a molecular level, of their shape selectivity [5]. A modulation of the steric hindrance of the grafted organometallic SnR3 complexes allows fine tuning of the separation properties of the zeolites modified by this method [5].
Similarly, (n-C4H9)2SnH2 reacts with vicinal silanol groups of partially dehydroxylated silicas with formation of the well-defined (SiO)2Sn(n-C4H9)2 surface complex (2) as shown in Eq. (2) [9].
| (2) |
Thermal treatment of 1 under vacuum leads, partly, to the stepwise formation (by a hydrogenolysis reaction with hydroxyl groups of the silica surface) of tin compounds in which tin is progressively linked to the silica surface via two, three and probably four oxygen atoms [10]: (SiO)2Sn(n-C4H9)2 (2) which can be prepared in a pure form (vide supra), (SiO)3Sn(n-C4H9) (3) and a fully dealkylated tin species (4). Simultaneously, tin(II) species are formed, probably by a β-H elimination (leading to an hydride species) followed by a reductive elimination of butane. As all these compounds are formed simultaneously, it is not possible to obtain a single well-defined complex at a given temperature. This phenomenon renders the mechanistic interpretation of the decomposition of 1 rather difficult. Since 2 is a possible intermediate in the thermal decomposition of 1, it was logical to prepare it by an independent route and to study the elementary steps of its transformation into less alkylated species.
Unfortunately, as always on divided surfaces, the structures of organometallic fragments on surfaces remain still hypothetical, at least to some extent. We have therefore decided to synthesize molecular models of 1, 2 and 3 with the silsesquioxane ligand [11] and to compare their spectroscopic properties to those of the grafted entities, in order to confirm the proposed structures. This ligand has the advantage of mimicking at a molecular level the local structure of crystobalite [11].
This paper reports both the study of the thermal transformation of 2 and the synthesis of these relevant molecular analogues. For this purpose, many methods such as volummetry, gas chromatography, elemental analysis, infrared spectroscopy, 13C CP–MAS and 119Sn MAS NMR were applied to the elucidation of the surface structure of a given species.
2 Experimental section
2.1 Materials and procedures
Aerosil silica (Degussa, 200 m2 g–1) was dehydroxylated under dynamic vacuum (10–4 Torr) for 14 h either at 200 °C (silica(200)) or at 500 °C (silica(500)).
Synthesis of the well-defined (SiO)2Sn(n-C4H9)2 (2) surface complex was achieved by reaction of (n-C4H9)2SnH2, at 25–100 °C, with the partially dehydroxylated silica [8]. 2 was characterized by elemental analysis of the solid (C4/Sn = 2), infrared spectroscopy (disappearance of the ν(Sn-H) band) and by 13C CP–MAS NMR spectroscopy (δ(α-C) = 20.0 ppm, δ(β-C) and δ(γ-C) = 26.5 ppm, δ(δ-C) = 13.0 and 11.3 ppm). The δ-carbon atoms showed two peaks in the 13C NMR spectra due to the fact that the terminal methyl group could interact or not with free silanol groups or siloxy bridges of the support (Fig. 1) as previously observed for Si–O–Sn(n-C4H9)3, for which this attribution was proved by numerous studies, for example as a function of the tin loading on the support. The structure of 2 was also deduced from the amount of hydrogen evolved during the grafting reaction.
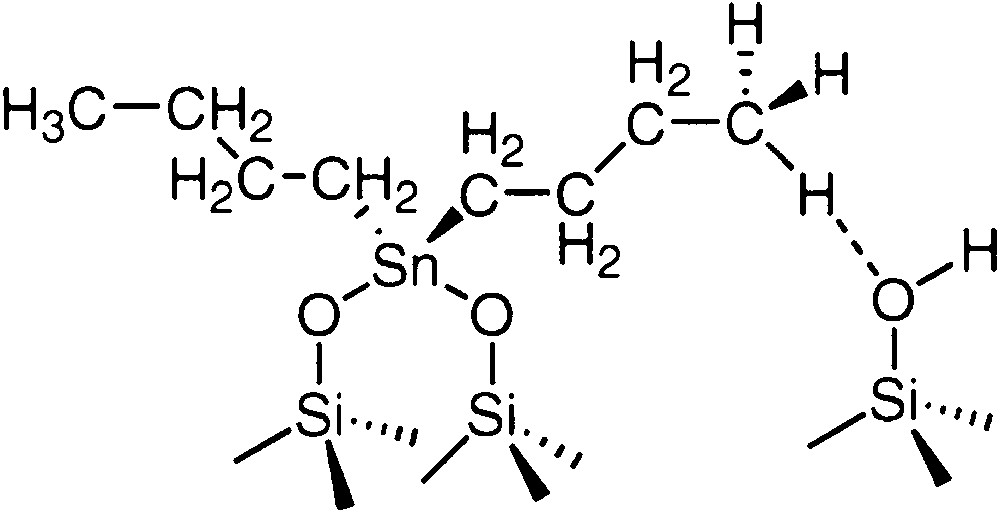
Proposed structure for 2 supported on silica.
Temperature-programmed decomposition experiments were performed under static vacuum (10–14 h). Before each temperature increment, a dynamic vacuum was made in the equipment (15 min, 25 °C, 10–4 Torr).
The evolution of the surface was followed by infrared spectroscopy and analysis of the evolved gases.
2.2 Equipment for analytical measurements
The experiments were carried out in a glassware equipment connected to a vacuum line and to a volumetric apparatus. The evolved gases (H2, C4H8, C4H10, etc) were analysed (qualitatively and quantitatively) by a volumetric technique and by gas chromatography.
2.3 13C CP–MAS NMR spectroscopy
The 13C CP-MAS NMR spectra were recorded on a Bruker MSL-300 spectrometer operating at 75.47 MHz. The probe head was a commercial double-tuned 7-mm double-bearing system from Bruker allowing spinning frequencies up to 4 kHz. The samples were introduced in the rotors made of zirconia under a dry nitrogen atmosphere in a glove box and tightly closed. Boil-off nitrogen was used for both bearing and driving the rotors. Under these conditions, no decomposition of the samples was observed during the course of the experiments as shown by 1H MAS NMR (same spectrum with the same intensities before and after recording the 13C CP–MAS NMR spectrum).
A typical cross-polarisation sequence was used: 90° rotation of the 1H magnetization (pulse length 6.2 μs), then contact between carbon and proton for 5 ms and finally recording of the spectrum under high-power decoupling. The delay between each scan was fixed at 5 s, to allow for the complete relaxation of the 1H nuclei (as proved by 1H NMR). The chemical shifts are given with respect to TMS by using adamantane as an external reference (δ = 37.7 ppm for the highest chemical shift). Sometimes, adamantane was directly added to the sample in the rotor in order to have an internal reference. No variation of the chemical shifts was detected compared to the use of an external reference.
2.4 Infrared spectroscopy
The infrared spectra were obtained on a Fourier transform Nicolet 10-MX spectrometer. The spectra were recorded in situ by using a special cell equipped with CaF2 windows.
The concentration of the butyl groups remaining on the surface as a function of the temperature was determined by the decay of the intensity of the ν(C–H) band at 2958 cm–1. The spectra were calibrated by using the harmonic bands of silica near 2000 cm–1.
2.5 Synthesis and characterization of molecular models
Except where noted, all experiments were carried out under an inert atmosphere of argon on a high-vacuum line applying Schlenk techniques. Bu3SnCl, Bu2SnCl2 and BuSnCl3 were purchased from Aldrich and were used without further purification. All solvents were dried and distilled under nitrogen by standard methods prior to use. Elemental analyses were performed by the ‘Service central d’analyses’ of the CNRS. In order to avoid precipitation of silicon carbide, the samples were heated at 1800 °C for analysis of carbon. The LSIMS mass spectral analyses were conducted on a Micromass ZAB2-SEQ spectrometer (caesium gun with acceleration of the Cs ions under 35 kV). The infrared spectra were recorded as pressed KBr pellets on a Nicolet 550 spectrometer. The liquid NMR spectra were recorded on a Bruker AM–300 (1H, 300 MHz; 13C, 75.46 MHz; 29Si, 59.62 MHz; 119Sn, 111.92 MHz) and on a Bruker CXP-100 spectrometer (1H, 100 MHz; 13C, 25 MHz). All chemical shifts are given relative to tetramethylsilane (1H, 13C and 29Si) and tetramethyltin for 119Sn. Classical sequences were used in all cases except for 29Si NMR spectra which were recorded with inverse-gated proton decoupling in order to minimize negative nuclear Overhauser enhancement effects and in presence of 0.02 M Cr(acac)3 to ensure complete relaxation between each scan.
In a first step, cyclopentyltrichlorosilane (c-C5H9SiCl3) was prepared by hydrosilylation of cyclopentene [12]. The incompletely condensed polyhedral oligosilsesquioxane [(c-C5H9)7Si7O9(OH)3)] was then obtained by the hydrolytic condensation reaction of c-C5H9SiCl3 in refluxing aqueous acetone, according to the previously described method [11].
2.5.1 Preparation of [(c-C5H9)7Si8O12Sn(n-C4H9)3(CH3)2] (1′)
In order to have one silanol left, the trisilanol [(c-C5H9)7Si7O9(OH)3)] was preliminary blocked on two silanol groups by reaction with Cl2SiMe2 in the presence of triethylamine. The reaction is quite the same than that of the trisilanol with dibutylchlorostannane and is performed in quite the same conditions [13]. Tributylchlorostannane (135 mg, 0.41 mmol, 0.9 eq.) was then slowly added to a solution of the blocked trisilanol (450 mg, 0.46 mmol) and triethylamine (233 mg, 2.30 mmol, 5 equiv) in n-hexane (20 ml) preliminary distilled over Na/K. The solution was stirred during 3 h at room temperature. After removing all volatiles by vacuum (10–4 Torr) at room temperature, the reaction product was extracted by n-hexane. The reaction is almost quantitative and the product is an oil which crystallises at –20 °C. 1′ was characterized by chemical analyses and 13C, 29Si and 119Sn NMR. 29Si {1H} NMR (59.62 MHz, C6D6, Cr(acac)3, 25 °C) δ –67.8, –66.4, –64.5, –64.4, –62.7 (s, 2: 2: 1: 1: 1) and –18.0 (s, 1). 13C {1H} NMR (75.46 MHz, C6D6, 25 °C) δ 13.8 (s, Cδ of butyl group), 16.5 (s, Cα of butyl group), 26.6 and 26.8 (s, Cβ and Cγ of butyl group), 1.0 and 1.09 ppm (s, 1:1, Si(CH3)), 22.9, 23.1, 23.7, 24.0, 26.2 (s, 1:1:2:2:1, CH of cyclopentyl ligand), 28.9, 28.2, 28.1, 28.0, 27.9, 27.6, 27.4, 27.3, 27.0, 26.9, 26.8 and 26.6 (s, CH2 of cyclopentyl ligand). 119Sn {1H} NMR (111.92 MHz, C6D6, 25 °C) δ 80.5 (s). Anal. calcd for C49H96SnO12Si8 (found): C, 48.21 (48.15); H, 7.92 (8.06); Sn, 9.72 (9.60) ; Si, 18.4 (18.81).
2.5.2 Preparation of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2′)
Dibutylchlorostannane (140 mg, 0.46 mmol) was slowly added to a solution of the trisilanol [(c-C5H9)7Si7O9(OH)3)] (403 mg, 0.46 mmol) and triethylamine (233 mg, 2.30 mmol) in tetrahydrofuran (30 ml). The solution, initially colourless, became turbidimmediately after addition of the first amount of Bu2SnCl2. It was stirred overnight at room temperature. Evaporation of the volatiles (25 °C, 10–2 Torr) followed by extraction with n-hexane and solvent removal in vacuo gave (2′) as an amorphous white foam. Crystallization at –30 °C in n-hexane afforded 316 mg (80% yield) as a colourless crystalline powder. 29Si {1H} NMR (59.62 MHz, C6D6, Cr(acac)3, 25 °C) δ –54.67, –60.70, –64.48, –65.39, –66.60 (s, 1:2:1:1:2). 13C {1H} NMR (75.46 MHz, C6D6, 25 °C) δ 14.06 (s, Cδ of butyl group), 22.49, 21.57 (s, Cα of butyl group), 27.44 and 27.21 (s, Cβ and Cγ of butyl group), 25.09, 23.70, 23.34, 23.22 (s, CH of cyclopentyl ligand), 28.96, 28.80, 28.48, 28.40, 28.26, 27.96, 27.89 (s, CH2 of cyclopentyl ligand). See also the results part for more data on this complex. 119Sn {1H} NMR (111.92 MHz, C6D6, 25 °C) δ –37.03 (s). 1H NMR (100 MHz, C6D6, 25 °C) δ 5.00 (vbr s, 1 H, OH), 1.94 to 0.78 (vbr m, 81 H, butyl and cyclopentyl groups). IR (KBr pellets): 3379 (m), 2947 (s), 2862 (s), 1449 (m), 1246 (m), 1099 (s), 1049 (s), 1007 (m), 939 (m), 912 (m), 882 (w), 503 (s) 422 (m) cm–1. Anal. calcd for C43H82SnO12Si7 (found): C, 46.63 (46.73); H, 7.41 (7.68); Sn, 10.72 (10.80). Molecular peak m/e = 1107 (MH+).
2.5.3 Preparation of [(c-C5H9)7Si7O12Sn(n-C4H9)] (3′)
A tetrahydrofuran solution (2 ml) of butyltrichlorostannane (161 mg, 0.57 mmol) was dropped to a solution of trisilanol [(c-C5H9)7Si7O9(OH)3)] (500 mg, 0.57 mmol) and triethylamine (288 mg, 2.85 mmol) in tetrahydrofuran (30 ml). The solution was stirred at 25 °C overnight and the product was extracted by n-hexane, as above. (3′) was obtained in quantitative yield. It was recrystallized at –30 °C in n-hexane and obtained as a colourless crystalline powder. 29Si {1H} NMR (59.62 MHz, C6D6, Cr(acac)3, 25 °C) δ –62.16, –65.16, –66.62 (s, 3: 1: 3). 13C {1H} NMR (75.46 MHz, C6D6, 25 °C) δ 13.30 (s, Cδ of butyl group), 19.14 (s, Cα of butyl group), 26.74 and 26.12 (s, Cβ and Cγ of butyl group), 24.17, 23.22, 22.92 (s, 3: 1: 3, CH of cyclopentyl ligand), 28.47, 28.02, 27.95, 27.62, 27.59, 27.27 (s, CH2 of cyclopentyl ligand). 119Sn {1H} NMR (111.92 MHz, C6D6, 25 °C) δ –169.48 (s). 1H NMR (100 MHz, C6D6, 25 °C) δ 1.73 and 1.30 (vbr m, butyl and cyclopentyl groups). IR (KBr pellets): 2953 (s), 2868 (s), 1564 (w), 1459 (m), 1248 (m), 1156 (s), 1077 (s), 1007 (m), 947 (m), 919 (m), 728 (w), 678 (w), 511 (s) 428 (m) cm–1. Anal. calcd for C39H72SnO12Si7 (found): C, 44.69 (43.8); H, 6.92 (6.90); Sn, 11.32 (11.5).
3 Results
We will first report the results on the thermolysis, under vacuum, of (SiO)2Sn(n-C4H9)2 (2) on two different silica samples dehydroxylated respectively at 200 (silica(200)) and 500 °C (silica(500)). We will then describe the synthesis and characterization of the model compounds.
3.1 Thermolysis under vacuum of (SiO)2Sn(n-C4H9)2 (2) supported on silica(200)
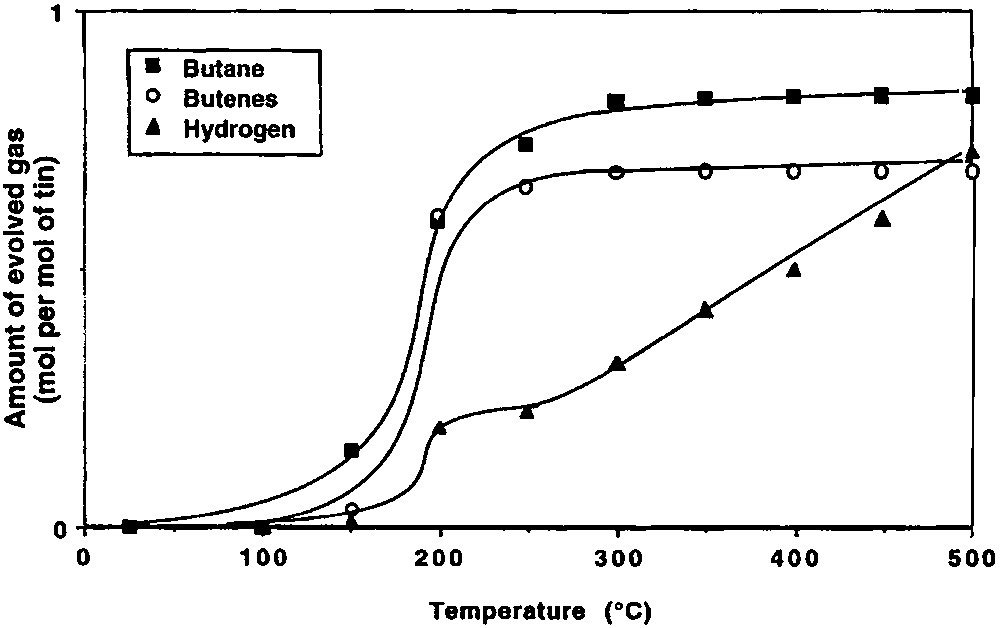
Fig. 2 shows the variation with temperature of the evolved gases during the thermolysis under vacuum of 2 grafted on silica(200). Different ranges of temperature can be considered.
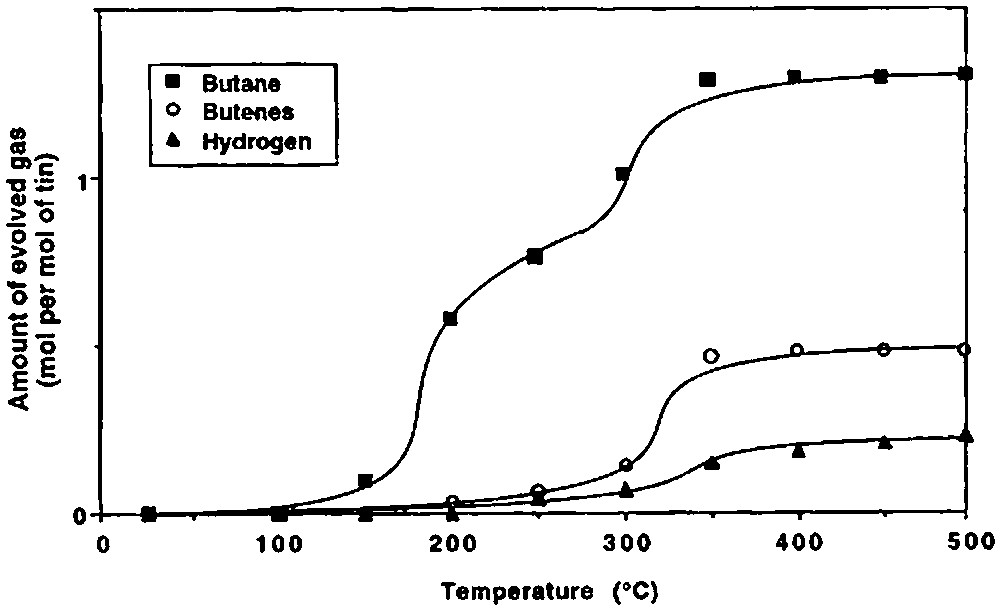
Gases evolved during the thermolyis, under vacuum, of 2 grafted on silica(200).
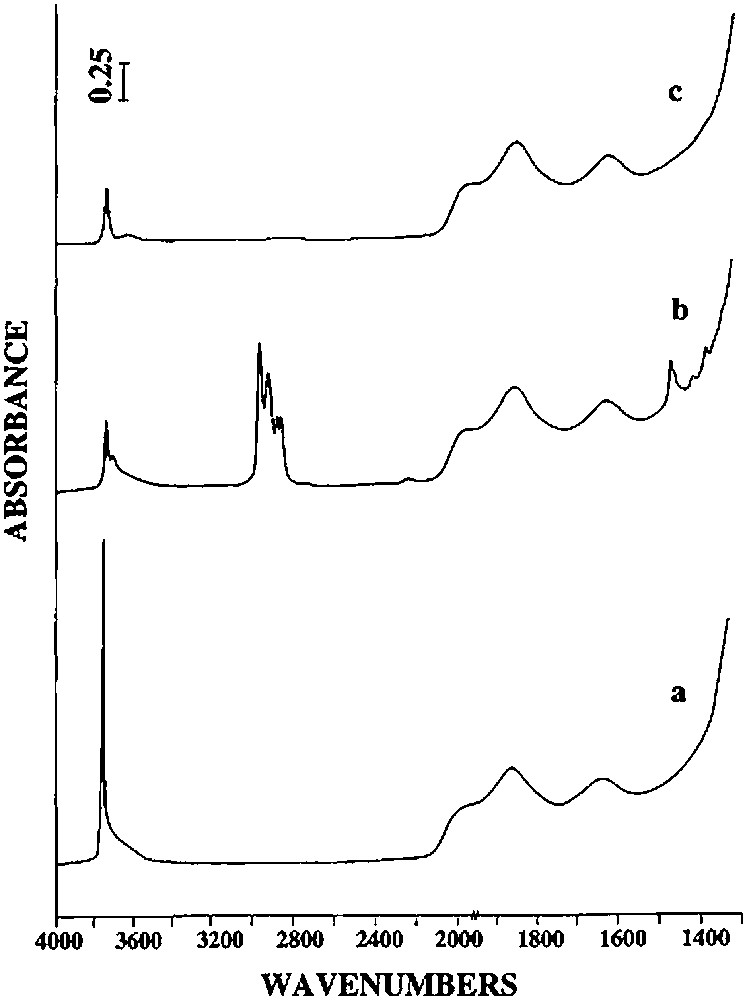
(1) Until 150 °C, almost no gas is evolved. No evolution of the infrared spectrum is observed, in particular in the ν(O–H), ν(C–H) and δ(C–H) vibrational bands regions (Fig. 3), indicating that 2 is thermally stable under these experimental conditions.
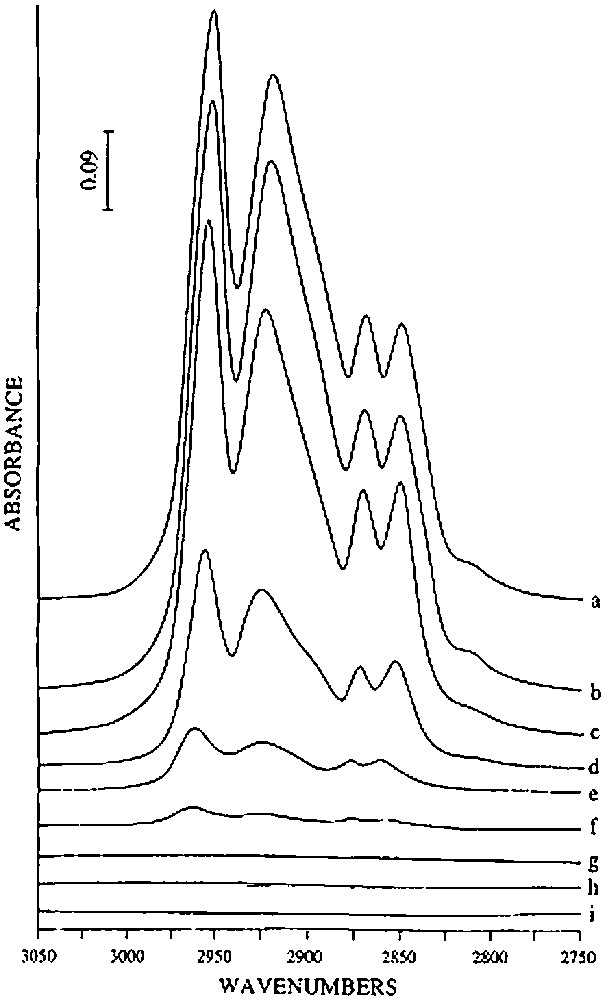
Evolution of the infrared spectrum, in the ν(C–H) spectral region, of (SiO)2Sn(n-C4H9)2 (2) grafted on silica(200) after treatment under vacuum at various temperatures: (a) untreated (b) 150 °C, (c) 200 °C, (d) 250 °C, (e) 300 °C, (f) 350 °C, (g) 400 °C, (h) 450 °C and (i) 500 °C.
(2) Between ca. 150 and 250 °C, 2 begins to react as evidenced by the evolution of n-butane (0.76 mole per mole of grafted tin), n-butene (0.06 mol) and hydrogen (0.04 mol). Simultaneously, the intensity of the ν(C–H) and δ(C–H) infrared bands decreases progressively (Fig. 3). A quantitative estimation of the elimination of butyl ligands can be made by measuring the decay of the intensity of the ν(C–H) band at 2958 cm–1: at 250 °C, ca. 1.2 butyl ligands per tin atom remain on the surface (Fig. 4). This result is consistent with the gas phase analysis that indicates the evolution of 0.82 mol of n-C4 hydrocarbons per mole of grafted tin.
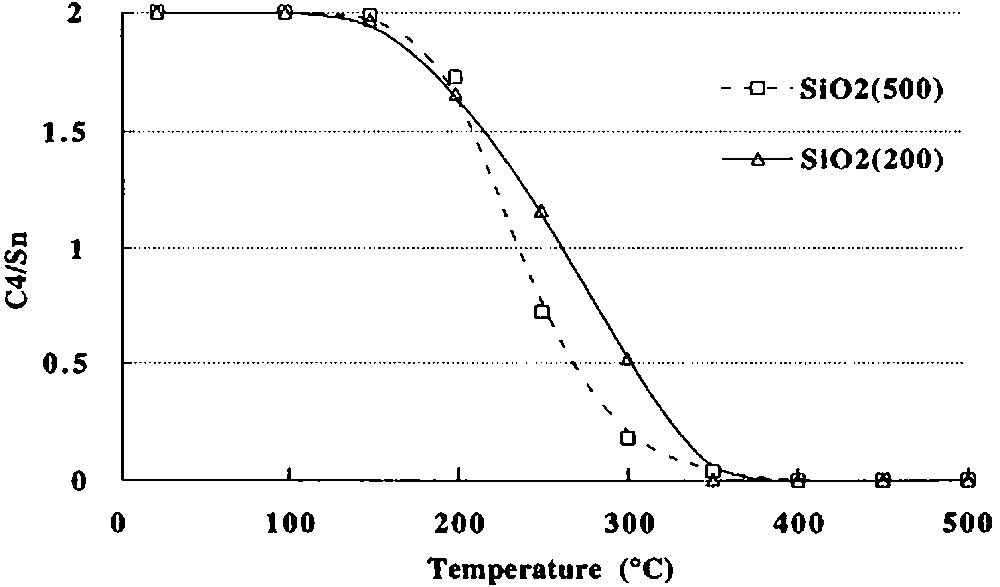
Number of C4 equivalents remaining per grafted tin as a function of temperature during the thermolysis, under vacuum, of (SiO)2Sn(n-C4H9)2 (2) grafted on (a) silica(200), (b) silica(500) (values deduced from the variation of the intensity of the ν(C–H) band at 2958 cm–1).
The 13C CP-MAS NMR spectrum of 2 shows peaks at 26.4 ppm (β-C and γ-C), 20 ppm (α-C), 13.0 and 11.3 ppm (δ-C in which the methyl group is free or develops an interaction with the surface respectively, see Fig. 1) (Table 1).
Comparison of 13C and 119Sn NMR chemical shifts of the surface species (SiO)1+xSnBu3–x and of the corresponding molecular models
| 13C NMR | 119Sn NMR | |||||||||
| Compound | Solution | CP–MAS | sol. | MAS | ||||||
| α | β | γ | δ | α | β | γ | δ | |||
| 1 b | 15.2 | 26.7 | 26.7 | 13.0 11.3a | 104 | |||||
| 1′ | 16.5 | 26.8 | 26.6 | 13.8 | 80.5 | |||||
| 2 c | 20.2 | 26.9 | 26.9 | 12.9 11.3a | ||||||
| 2′ | 23.7 | 27.5 | 27.2 | 14.1 | 22.8 | 26.9 | 26.9 | 13.3 | –37 | –36.5 |
| 3 d | ||||||||||
| 3′ | 19.1 | 26.2 | 26.1 | 13.3 | 23.8 | 27.3 | 27.3 | 12.9 | –169.5 | –198.9 |
a Terminal methyl group hydrogen bonding with adjacent silanol group.
b Obtained by reaction of Bu3SnH with SiO2(500).
c Obtained by reaction of Bu2SnH2 with SiO2(500).
d Obtained by thermolysis of (SiO)2SnBu2.
After heating at 200 °C during 14 h, the NMR spectrum is only slightly modified: (i) the relative intensity of the peak at 13.0 ppm (free methyl group) vs. that at 11.3 ppm (methyl group interacting with the oxygen atom of an hydroxyl bond of the surface) has decreased, in agreement with the assumption that the smaller the number of butyl ligands on the metal, the higher the number of hydrogen bonded terminal methyl groups; (ii) a new broad signal appears at ca. 21 ppm, as evidenced by the deconvolution of the spectra. From previous data obtained in the case of the thermolysis of Si–O–Sn(n-C4H9)3 (1) [10], this peak can be ascribed to the α-C carbon atom of a mono-alkylated species, in which the tin atom is linked to the silica surface by three oxygen atoms (SiO)3Sn(n-C4H9) (3). Note also that the replacement of a butyl ligand by a Si–O– group has a non-negligible effect only on the downfield chemical shift of the α-C carbon atom (ca. 4 ppm). One can also point out that, in molecular chemistry, the 13C NMR peak of the α-C carbon atom of an alkyl tin chain is shifted towards low fields when alkyl ligands are substituted by alkoxy ones [14].
(3) Between 250 and 350 °C, 0.54 mol of n-butane, 0.43 mol of n-butene and 0.11 mol of hydrogen are evolved per mole of grafted tin. The intensities of the ν(C–H) and δ(C–H) bands continue to decrease; these bands disappear after heating at 350 °C (Figs. 3 and 4). The ν(O–H) band at 3698 cm–1, which was attributed to hydroxyl groups interacting with the terminal methyl groups of the butyl ligands also disappears, in agreement with these results.
The 13C CP–MAS NMR spectrum of a sample treated at 300 °C exhibits similar peaks than those already discussed for the sample heated at 200 °C. The ratio of the intensities of the two peaks at 13.0 and 11.3 ppm continues to decrease, in parallel with the resonance at 20 ppm (attributed to the α-C carbon atom of 2). So, after treatment at 300 °C, solid-state NMR shows that a small amount of 2 remains on the silica surface, in coexistence with 3 and also probably some fully dealkylated tin species. The reason for the discrepancy between the infrared and NMR results is probably related to the different amounts of solid used for these two sets of experiments (~10 mg for infrared spectroscopy and ~1 g for solid-state NMR).
(4) Between 350 and 500 °C, only small amounts of hydrogen are evolved (0.08 mole per mole of grafted tin).
At the end of the thermal treatment, 1.3 mol of n-butane, 0.49 mol of n-butene and 0.23 mol of hydrogen have been evolved per mol of grafted tin. The remaining carbon, probably as coke as it can be expected from the dark colour of the sample (and corresponding to 0.21 mol of C4), is found on the surface by microanalysis (wt.% C = 0.19; wt% Sn = 2.20; C/Sn = 0.85). Note also that during all steps of the thermolysis of 2 only n-butane and n-butene are evolved as hydrocarbons.
3.2 Thermolysis under vacuum of (SiO)2Sn(n-C4H9)2 (2) supported on silica(500)
When the initial dehydroxylation step of the silica is conducted at 500 °C, the relative amounts of evolved gases are quite different from those detected in the case of silica(200) (Fig. 5).
Gases evolved during the thermolysis, under vacuum, of 2 grafted on silica(500).
On silica(500), n-butene and hydrogen are formed at more moderate temperatures than on silica(200). Between 100 and 300 °C, 0.83 mol of n-butane, 0.69 mol of n-butene and 0.32 mol of hydrogen are evolved per mole of grafted tin. Between 300 and 500 °C, only hydrogen (0.41 mole per mole of grafted tin) is formed. Infrared spectroscopy shows that the intensity of the ν(C–H) and δ(C–H) bands decreases as a function of temperature, as for silica(200) (Fig. 3). Simultaneously, and as already observed on silica(200), the intensity of the ν(O–H) band at 3698 cm–1 due to silanol groups interacting with the terminal methyl groups decreases. When no organic ligands remain on the surface, this band has completely disappeared. After heating at 500 °C, the intensity of the ν(O–H) band at 3747 cm–1, characteristic of free Si–OH entities, is clearly weaker than on the initial silica(500) (Fig. 6). This result suggests that no migration of tin atoms occurs during the thermolysis. If such migration had occurred significantly, one would expect that some silanol groups should be regenerated.
Infrared spectra of (a) silica(500), (b) (SiO)2Sn(n-C4H9)2 (2) supported on silica(500), (c) (SiO)2Sn(n-C4H9)2 (2) treated under vacuum, at 500 °C.
As on silica(200), no hydrocarbon other than n-butane and n-butene evolved during the thermolysis and the remaining carbon is found, probably as coke, on the surface (wt.% C = 0.29; wt.% Sn = 1.77; C/Sn = 1.6).
3.3 Synthesis and characterization of molecular models
Our purpose was to synthesize the molecular models of the starting organometallic fragments SiOSn(n-C4H9)3 (1) and (SiO)2Sn(n-C4H9)2 (2) as well as their decomposition product (SiO)3Sn(n-C4H9) (3), where the tin atom is surrounded respectively by one, two, or three butyl ligands and three, two or one siloxy groups. We used the trisilanol [(c-C5H9)7Si7O9(OH)3)] as model of the silica surface [11] and we adapted the synthesis of a tin derivative from a similar silsesquioxane, previously reported by Feher et al. [15]. So, the reaction of equimolar amounts of the trisilanol and Bu2SnCl2 or BuSnCl3 in the presence of an excess of triethylamine led to the quantitative formation of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2′) and [(c-C5H9)7Si7O12Sn(n-C4H9)] (3′). For the synthesis of a molecular model of (1), we used a trisilanol firstly blocked with Si(CH3)2Cl2 in order to have only one hydroxyl group left. The reaction of the latter with Bu3SnCl yielded [(c-C5H9)7Si8O12Sn(n-C4H9)3(CH3)2] (1′). The different reactions are summarized in Fig. 7.
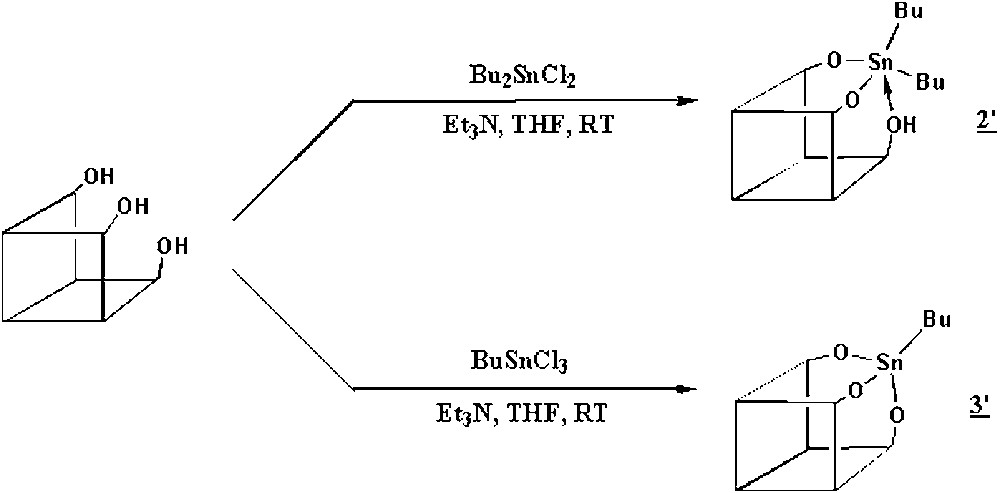
Reaction scheme for the synthesis of molecular models.
These compounds were characterized by various analytical and spectroscopic methods. Unfortunately, attempts to obtain crystals of sufficient good quality for solving the structure by X-ray diffraction failed. Indeed the crystals grown along one dimension, rendering the obtention of data of good quality very problematic. However, NMR spectroscopy proved to be very useful for the determination of the structures of (1′), (2′) and (3′).
Chemical analysis confirms the formula proposed for compound (3'), in agreement with one butyl ligand and one silsesquioxane fragment per tin atom. The 13C and 29Si NMR data show clearly that the complex had the C3v symmetry group (three peaks in the 29Si NMR spectrum of relative intensities 3:1:3 and three peaks, with the same intensity ratio, for the CH atoms of the cyclopentyl rings in the 13C NMR spectrum). The 13C NMR spectrum shows also resonances at 13.3, 19.1, 26.1 and 26.2 ppm attributed respectively to the Cδ, Cα, Cβ and Cγ carbon atoms of the butyl ligand. Compared to BuSnCl3, the greatest variation is obtained for the Cα carbon atom (from 32.42 ppm to 19.14 ppm). In agreement with the literature data, the other resonances are only slightly shifted. 119Sn NMR gives a peak at –168.5 ppm, consistent with a four-coordinated tin atom in a (RO)3SnR complex [16].
We can deduce from these data that in 3' the tin atom is tetrahedrally coordinated to three siloxy bridges of the silsesquioxane and to one butyl group, as shown in Fig. 7. Such a structure corresponds well to what is expected for compound 3.
The chemical analysis and the 29Si, 13C and 119Sn NMR spectra of compound 1' are in agreement with the expected structure. The 13C NMR spectrum shows, in the region of the CH groups of the cyclopentyl ligands, five peaks at 22.9, 23.1, 23.7, 24.0 and 26.2 ppm (relative intensities 1:1:2:2:1), showing that 1′ has a Cs symmetry group. The 29Si NMR spectrum is also in agreement with this symmetry (presence of five peaks in a 2:2:1:1:1 relative intensity for the silicon atoms bound to the cyclopentyl groups). The 13C NMR spectrum shows also two resonances at 1.1 and 1.3 ppm (methyl groups of SiMe2) and peaks at 13.8, 16.5, 26.6, and 26.8 ppm attributed respectively to the δ, α, β and γ carbon atoms of the butyl ligands. The presence of only one peak for each of the carbon atoms of the three butyl ligands while the symmetry is Cs is probably related to a rapid rotation of the –SnBu3 fragment around the Sn–O bond, resulting then in apparent equivalence of the three organic groups. The 119Sn NMR spectrum shows only one resonance at 80.5 ppm, consistent with a four-coordinated tin atom as in a (RO)SnR′3 compound [16]. As a consequence, we assign tentatively a tetrahedral structure to the tin atom in complex (1′), with tin linked by one siloxy group to the silsesquioxane.
Similarly, chemical analysis of compound 2′ is in agreement with two butyl groups and one silsesquioxane entity per tin atom. Mass spectrometry agrees also with this result (molecular peak at m/e = 1107 (for MH+)). The 29Si NMR spectrum shows that the complex has a symmetry group lower than C3v. Indeed, the 29Si NMR spectrum presents five peaks in a 2:1:1:2:1 intensity ratio. It is relatively difficult to determine, from these data, if the two butyl units are not equivalent at room temperature, as it is the case for the two methyl groups of the Si(CH3)2 moiety in the 1′ complex. We have then undertaken a more detailed study of this complex by proton and carbon-13 NMR. As the methyl group of the butyl ligands gives a sharp signal, we have first performed a 1D TOCSY from this methyl group. Depending on the mixing time, the various protons of the butyl chain appeared then as shown in Fig. 8 The protons on the γ carbon give a peak at ca. 1.35 ppm, those on the β carbon atom a peak at ca. 1.70 ppm and those on the α carbon atom two peaks at ca. 1.45 and 1.60 ppm. By using the TOCSY transfer from the methyl (mixing time 84 ms) and a HSQC gradient, a correlation could then be obtained between the proton and carbon signals (Fig. 9). These results allow us to conclude unambiguously that the α carbon atom of the butyl group gives two peaks at 21.57 and 22.49 ppm, the β-carbon atom two peaks at 27.21 and 27.44 ppm, the γ-carbon atom one peak at 27.44 ppm and the δ-carbon atom one peak at 14.06 ppm. As a consequence, the two butyl chains are not equivalent at room temperature.
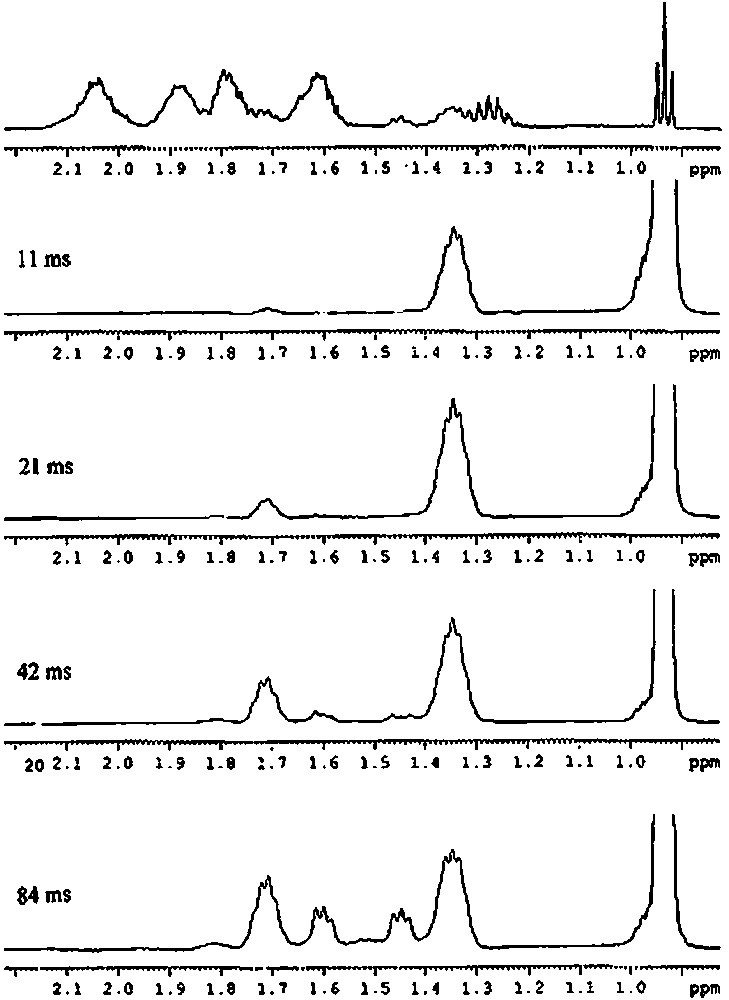
1H 1D TOCSY solution NMR spectra of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2′) (solvent: toluene) as a function of the mixing time.
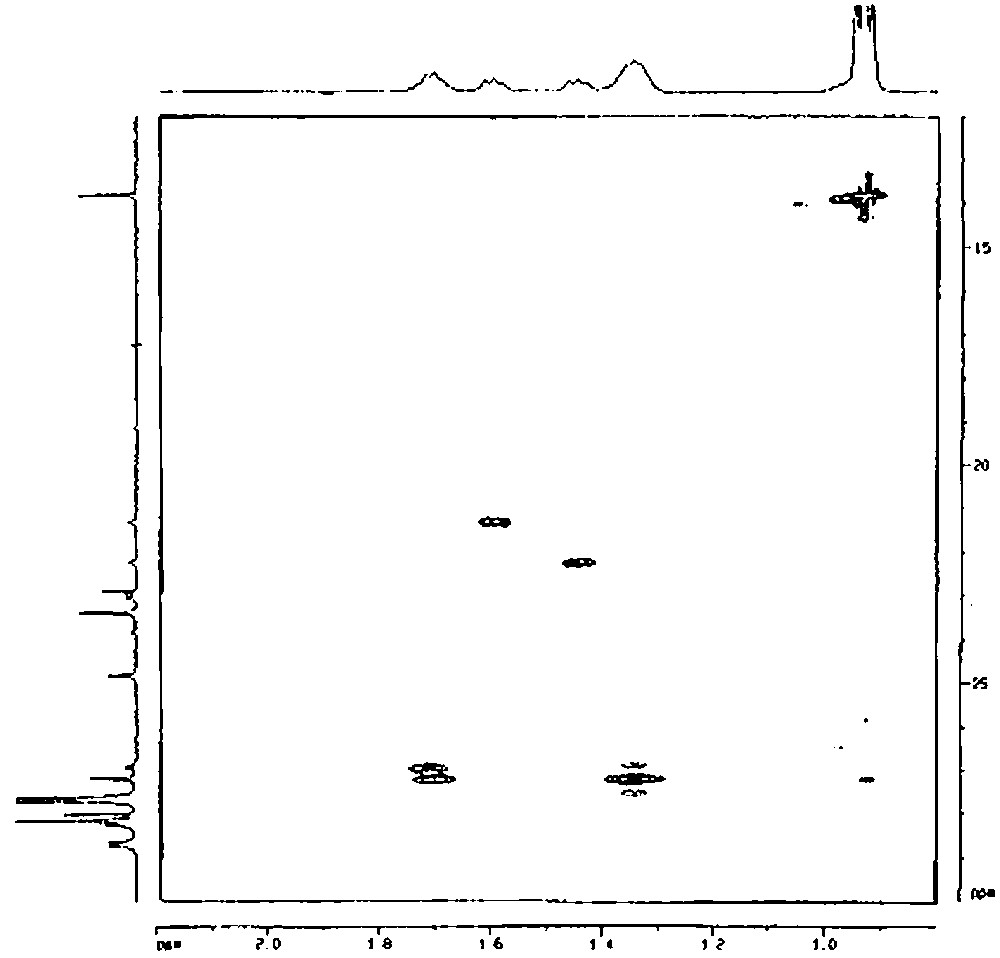
2D 1H–13C TOCSY-HSQC of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2′) (solvent: toluene).
The main problem that subsists is the coordination or not of the remaining hydroxyl group to the tin atom. Infrared spectroscopy, which is able to distinguish between free and H-bonded silanols, is not useful in this case as ν(O–H) frequency corresponds to H-bonded silanols, but the same shift is observed on silica when they interact with the alkyl fragments of the organometallic fragment. 119Sn NMR gives a peak at –37 ppm, which could be consistent with a four-coordinated tin atom in a (RO)2SnR2 complex [14,17,18]. However, these data are not sufficient to exclude the coordination of the hydroxyl group. Indeed, a weak intramolecular dative bonding between a hydroxyl group and tin has only a little effect on the tin chemical shift [19]. It is possible to conclude by studying the possibility of an exchange between the two butyl groups. Indeed, when the tin atom is tetracoordinated, it is necessary to break Sn–C bonds in order to exchange the two butyl groups, leading to a mechanism with a highly energetic transition state. In contrast, if the tin atom is pentacoordinated, the fifth ligand being the remaining hydroxyl group of the silsesquioxane, a pseudorotation can occur. Such an Sn–O intramolecular coordination, leading to a pentacoordination, has been reported for tetraorganotin derivatives that were previously regarded as unable to show a pentacoordinate structure [19,20]. By recording the 2D ROESY spectrum of 2′, the presence of an exchange can be emphasized by the presence of cross-peaks between the two α-carbon atoms. As shown in Fig. 10, these peaks are well seen, probing the exchange, even if it is very slow.
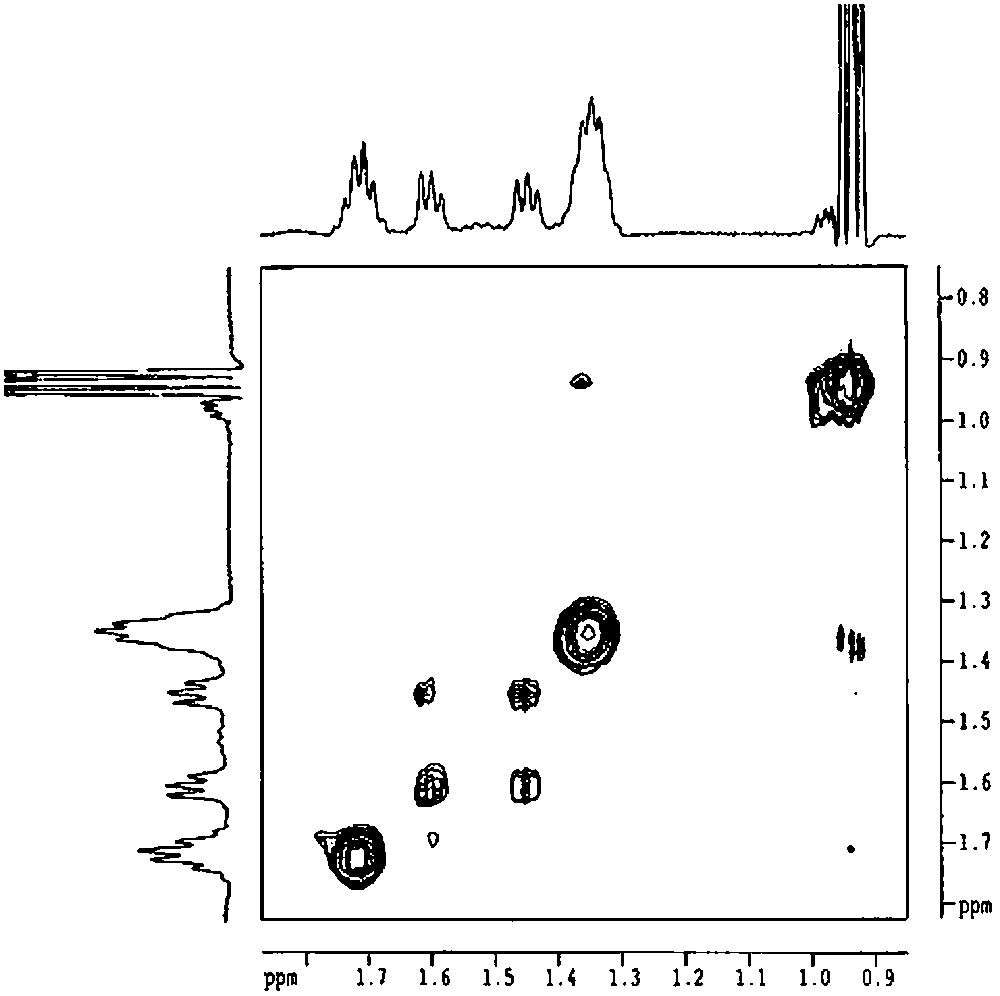
2D ROESY spectrum of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2') (solvent: toluene).
Moreover, In order to comfort this hypothesis and to prove the existence of the pseudorotation mechanism, we have recorded the 13C NMR spectrum of 2′ at low temperature. A reversible splitting of the four resonances of the butyl groups is then observed, showing that at low temperature the two butyl ligands are not equivalent. As an example, Fig. 11 shows the variation of the 13C NMR spectrum in the region of the methyl groups of the butyl ligands as a function of the temperature. The activation energy of the exchange process is found to be about 13 kcal mol–1 and corresponds well to what is expected for a pseudorotation.

Variable-temperature 13C NMR spectra of [(c-C5H9)7Si7O11(OH)Sn(n-C4H9)2] (2') (solvent: toluene).
In conclusion, we can say that in 2′ the tin atom is in a trigonal bipyramidal environment with a pseudorotational process that interconverts the butyl groups. Such a structure is in complete agreement with what can be expected from molecular modelling.
4 Discussion
The above results can be rationalized in terms of surface structures and mechanisms of interconversion of surface organometallic fragments. The surface structures of 1, 2 and 3 seem to be well understood on silica. 1, which has been published previously [8] has 13C and 119Sn NMR spectra quite similar to those of 1′ (Table 1) showing that 1′ is a good model of this surface species. 2′ has approximately the same chemical shifts in solution and in the solid state (Table 1), showing that there is no unusual packing or oligomeric effects in the solid state. We were never successful in obtaining a solid-state 119Sn NMR spectrum of the surface complex 2. A reason for this may be found in the solid-state 119Sn NMR spectrum of 2′, where we observed many spinning sidebands, due to the great chemical shift anisotropy around tin. This effect, if present in 2, would broaden the already very weak signal into the baseline. However, in the butyl region of the 13C NMR spectrum, a good correlation is observed between the two solid-state NMR spectra of 2 and 2′,even if one should note that:
- • a very small difference in the C(α) chemical shifts, likely due to an additional oxygen atom in the coordination sphere of 2′;
- • in the spectrum of 2 we observed an hydrogen bonding between the terminal methyl of the butyl group and the silanol groups of the surface that produced a new resonance at 11.3 ppm; no such effect was observed for the molecular model perhaps due to the lack of a sufficient ‘silica-like’ periphery.
More specifically, (2′) can be considered as a model of the first step of the hydrolysis reaction as it contains a hydroxyl group in slight interaction with the tin atom. Another proof of this was found during the thermolysis of the molecular compounds: After heating at 200 °C, small amounts of butane and butene (ca. 0.1 molecule per tin) were found, as for the surface species, showing that the decomposition passes probably through the same routes, even if, in the case of the molecular models, the reactivity was smaller, due to the less acidic character of the hydroxyl groups.
The solution and solid-state 119Sn NMR spectra of 3′ are quite different from each other (Table 1): a variation of 30 ppm is noted and, as in 2′, the solid-state spectrum shows a high degree of chemical-shift anisotropy of the tin atom. This could indicate a packing effect that increases the coordination number of the tin atom in the solid state. There is also a downfield shift of the α-carbon atom between the solution and solid-state spectra (from 19.1 to 23.8 ppm) also consistent with additional oxygen coordination for 3′ compared to 2′. As for 2, we were never able to obtain a 119Sn NMR spectrum of complex 3, in agreement with the presence of numerous spinning sidebands in the spectrum of 3′. As above, no indication of a methyl group in interaction with hydroxyl groups was found, but in this case it was not surprising, as there are no silanol groups remaining in the complex.
The molecular compounds 1′, 2′, and 3′ can so be considered as good models of the surface species 1, 2 and 3. For 2, the molecular model 2′ seems to indicate that a silanol group could be coordinated to the SnIV complex; this is probably occurring to a greater extent on silica(200) than on silica(500).
Species 3 has never been obtained in a pure form since the decomposition pathways are not fully selective but its existence has been seen among other species by 13C CP-MAS NMR. The synthesis of 3′ is also a strong support for the existence of such a surface organometallic structure. Dealkylated tin(II) and (IV) atoms linked to 2 or 4 surface oxygen atoms have already been proved, especially by Mössbauer [10].
Regarding the thermal transformation of 2 on the surface of silica(200) or silica(500), Fig. 12 summarizes, from the above data, two different decomposition pathways. When the surface is rich in hydroxyl groups (silica(200)), 2 might be pentacoordinated like in 2′ and the easiest reaction is the hydrolysis of one butyl group leading to 3 with evolution of butane. This path is consistent with the results of Fig. 2. When the surface is poor in hydroxyl groups (silica(500)), 2 has more likely a tetracoordinated structure with two butyl groups and two oxygen atoms of the surface. There is no hydroxyl group in the vicinity of tin and the only decomposition pathway is a dismutation pathway leading to butane and butene in equivalent amounts. This is consistent with the results of Fig. 5.
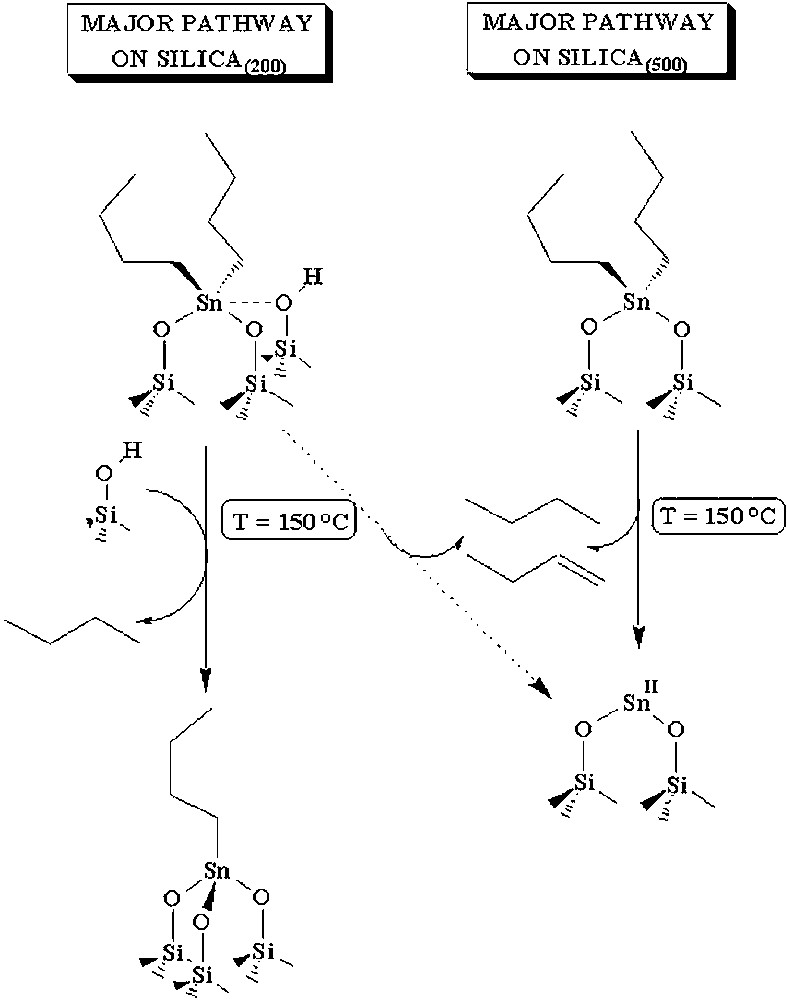
Decomposition pathways of (SiO)2Sn(n-C4H9)2 on silica dehydroxylated at 200 or 500 °C.
There is no clear cut between the two mechanisms and this is the reason why after treatment at high temperature, there is always a mixture of the two decomposition products, even if one route is predominant.
5 Conclusion
We have shown that the proposed structures for the surface species (SiO)1+xSn(n-C4H9)3–x (x = 0, 1, 2) formed during the thermolysis of alkyl tin complexes grafted on the surface of silica were reasonable, the spectroscopic data being in good agreement with those of molecular models. We have also shown that the decomposition mechanism under vacuum is the same for all compounds and passes through two different mechanisms depending on the density of hydroxyl groups on the support.
Acknowledgements
The authors wish to acknowledge Dr G.N. Niccolai for helpful discussions.