

1 Introduction
Among the carbonylation reactions, hydroformylation has been playing an outstanding role for some decades [1]. Several phosphorus containing mono- and diphosphines, phospholes, and heterobidentate ligands have already been tested in cobalt, rhodium and platinum-catalysed hydroformylation reactions and have been referenced recently [2]. Both the hydroformylation of simple substrates like propene and the asymmetric hydroformylation (e.g., that of vinylaromatics resulting in α-phenylpropanals, the precursors of α-aryl-propionic acids, which are of pharmacological importance [3]) hold an enormous potential in synthesis. Due to their high practical importance, hydroformylation catalysts have received particular attention.
The synthesis of esters from alkenes via carbonylation reactions has also a potential in synthetic organic chemistry. In the hydroalkoxycarbonylation (‘hydroesterification’) reaction, carbon monoxide and an alcohol are added to an alkene [4]. The reaction has an obvious relation to hydroformylation, where alcohol is used instead of hydrogen. Hydroalkoxycarbonylation of a linear olefin resulted in the predominant formation of the linear ester, both in the presence of palladium [5] and platinum [6] bimetallic catalysts.
A double hydroalkoxycarbonylation of conjugated dienes like butadiene provides a direct route to diesters, like dialkyl adipates [7].
In spite of examples aimed at the synthesis of more complicated structures of biological importance [8–13], the palladium-catalysed hydroalkoxycarbonylation of simple vinyl aromatics like styrene still remains the most important reaction. These compounds may serve not only as model derivatives, but the predominantly formed branched esters containing a stereogenic centre in position α can be considered as useful intermediates. Therefore, the enantioselective hydroalkoxycarbonylation is of special interest [14,15].
To find an appropriate balance of chemo- and regioselectivities, as well as a satisfactory catalytic activity, the search for efficient catalysts is a never-ending story. In the present work, we describe the application of novel phosphorus ligands either with an endo- or with an exocyclic phosphorus atom [16,17] in the rhodium-catalysed hydroformylation and in the palladium-catalysed hydroalkoxycarbonylation of styrene. Our systematic work on the fine tuning of the steric and electronic properties of the phosphole ligands [18] made available novel cyclic P-ligands, as well as ligands possessing a cyclopropane moiety (Fig. 1).
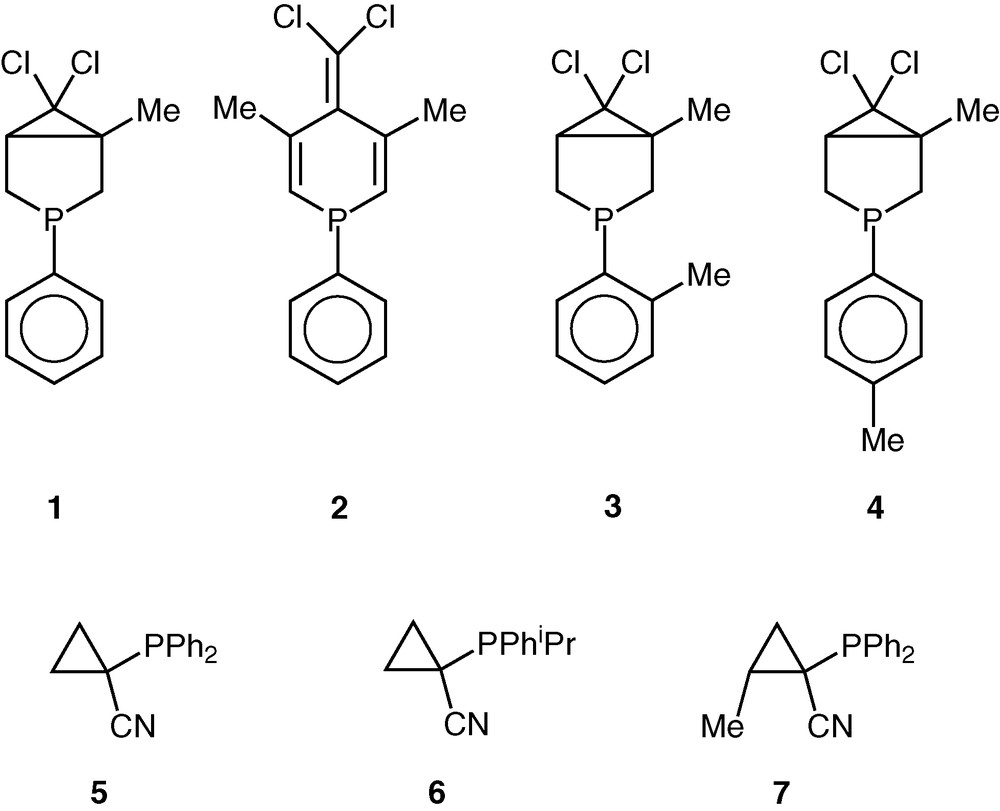
Compounds 1 to 7.
2 Results and discussion
The most active complexes used in hydroformylations have the general formula HxMy(CO)zLn, where M = cobalt and rhodium. We wished to test the activity of Rh(III) complexes lacking a CO moiety in the coordination sphere. It was also a challenge for us to investigate the properties of catalysts with novel P-ligands.
Styrene (8) displaying a relatively high reactivity among the alkenes was chosen as the model substrate that was reacted with CO/H2 (1/1) at 40–100 °C and at a pressure of 100 bar (Table 1) in the presence of one of the RhCl2(Cp*)(L)-type precursors (where L = 1–7, Fig. 1).
| (1) |
Hydroformylation (HF) and hydroethoxycarbonylation (HEC) of styrene with rhodium–Cp*–(phosphorus ligand) and palladium–(phosphorus ligand) (ligand = 1–7) complexes, respectively a
| Entry | Complex | Reaction | Temp. (°C) | Reaction time (h) | Conv. (%) | TOFe(h–1) | Rc b (%) | Rbr c (%) |
| 1 | RhCl2(Cp*)(1) | HF | 100 | 12 | 19 | 63 | 96 | 85 |
| 2 | RhCl2(Cp*)(1) | HF | 70 | 24 | 28 | 40 | > 99.5 | 86 |
| 3 | RhCl2(Cp*)(2) | HF | 100 | 11 | 25 | 91 | 98 | 83 |
| 4 | RhCl2(Cp*)(2) | HF | 70 | 24 | 2 | 3 | > 99.5 | 95 |
| 5 | RhCl2(Cp*)(2) | HF | 40 | 22 | 0 | — | — | — |
| 6 | RhCl2(Cp*)(3) | HF | 40 | 14 | 0 | — | — | — |
| 7 | RhCl2(Cp*)(3) | HF | 100 | 4 | > 99.5 | 1000 | > 99.5 | 33 |
| 8 | RhCl2(Cp*)(3) | HF | 40 | 72 | 0 | — | — | — |
| 9 | RhCl2(Cp*)(4) | HF | 100 | 4 | 99.3 | 993 | > 99.5 | 42 |
| 10 | RhCl2(Cp*)(4) | HF | 40 | 70 | 0 | — | — | — |
| 11 | RhCl2(Cp*)(5) | HF | 100 | 9 | 97 | 431 | > 99.5 | 68 |
| 12 | RhCl2(Cp*)(5) | HF | 70 | 24 | > 99 | 167 | > 99.5 | 27 |
| 13 | RhCl2(Cp*)(5) | HF | 40 | 16 | 0 | — | — | — |
| 14 | RhCl2(Cp*)(5) | HF | 40 | 26 | 2 | 3 | > 99.5 | 39 |
| 15 | RhCl2(Cp*)(6) | HF | 100 | 4 | 99.5 | 995 | > 99.5 | 34 |
| 16 | RhCl2(Cp*)(6) | HF | 40 | 70 | 52.5 | 30 | > 99.5 | 5 |
| 17 | RhCl2(Cp*)(7) | HF | 100 | 4 | 15 | 150 | > 99.5 | 24 |
| 18 | RhCl2(Cp*)(7) | HF | 40 | 70 | 0 | — | — | — |
| 19 | trans-PdCl2(2)2 | HEC | 90 | 48 | 2 | 2 | > 99 d | >99 |
| 20 | trans-PdCl2(5)2 | HEC | 90 | 48 | 21 | 17 | > 99 d | 98 |
a Reaction conditions: 100 bar CO/H2 = 1/1; Rh/styrene = 1:4000; solvent: toluene (HF); 90 bar CO; Pd/styrene = 1:4000; solvent: toluene/ethanol (HEC).
b Chemoselectivity (9+10)/(9+10+11) × 100 (HF); (12+13)/(12+13+oligomers) × 100 (HEC).
c Regioselectivity towards branched regioisomer 9/(9+10)*100 (HF); 12/(12+13) × 100 (HEC).
d Oligomerisation (dimerisation) of styrene as side reaction.
e Turnover frequency: mmol converted substrate/(mmol catalyst × reaction time).
In addition to the formyl regioisomers 9 and 10, some hydrogenation product 11 is also expected. Surprisingly, by using RhCl2(Cp*)(ligand) catalytic precursors, the formation of 11 was negligible (less than 0.5% and, in most cases, less than the detection limit).
The hydroformylation activity of RhCl2(Cp*)(1) and RhCl2(Cp*)(2) catalysts is moderate and the conversions are nearly the same (Table 1, entries 1 and 3). Higher activities have been observed with the cyclopropyl ligands 5 and 6 bearing an exocyclic phosphorus atom (Table 1, entries 11 and 15). Moreover, for longer reaction times, the latter ligand proved to be active even at 40 °C (Table 1, entry 16). Note that the introduction of a methyl substituent in the cyclopropane ring in trans position to the phosphorus atom (ligand 7) resulted in a spectacular decrease of the catalytic activity (Table 1, entries 11 and 17).
The catalysts formed with ligands possessing o-tolyl and p-tolyl substituents (3 and 4) can be considered as inactive at low temperatures, but the rhodium-Cp* type complexes of both ligands provided excellent conversions at 100 °C in comparison with the catalyst containing ligand 1 with similar structure, but bearing a phenyl group at the phosphorus atom (Table 1, entries 6–10). Thus, the introduction of tolyl groups instead of the phenyl one caused an unexpectedly high increase in activity.
It is worth noting that the chemoselectivity of the hydroformylations is excellent in all cases, where the catalyst is active at all. Ethylbenzene could be detected by immediate GC–MS analysis only in traces (vide supra).
As far as the regioselectivity is concerned, a dramatic drop in regioselectivity has been observed in the ligand series 1, 3, 4 going from 1 to 3–4. The introduction of o- and p-tolyl substituents (3 and 4) caused a pronounced decrease of the regioselectivity from 85% to 33% and 42%, respectively (Table 1, entries 1, 7 and 9).
A further feature has been observed by the application of the cyclopropane-based ligands 5 and 6, i.e. the regioselectivity towards the branched aldehyde (9) seems to be significantly influenced by the basicity of the phosphorus ligands. The use of the more basic phosphine 6 results in lower regioselectivities. Moreover, the selectivity changes in favour of the linear aldehyde (10) (Table 1, entries 11 and 15, respectively). Going from ligand 5 to ligand 7, the regioselectivity again decreased (Table 1, entries 11 and 17). It is noteworthy that the activity and the regioselectivity of the hydroformylation in the presence of 5–7 are changing in the opposite direction.
It has been shown that the lower the temperature was, the lower regioselectivities towards the branched aldehyde 9 were detected, i.e. a decrease in temperature favours the formation of the linear aldehyde 10 (Table 1, entries 11 and 12). This tendency is opposite to that observed with well-known preformed (e.g., HRh(CO)(PPh3)3) or in situ prepared rhodium–PPh3 systems [19]. Similar regioselectivity-temperature dependence has been observed for the analogous HRh(CO)(dppe) system.
The application of widely used in situ systems formed from [Rh(COD)Cl]2 or [Rh(CO)2Cl]2 precursors in the presence of conventional PPh3 or α,ω-bis(diphenylphosphino)alkanes resulted in regioselectivities towards 9 up to 96% [20].
Considering the whole ligand series (1–7) investigated, it turned out that using the catalytic system containing sterically slightly more bulky (and more basic) ligands (3, 4, and 6) the activity increases, while the regioselectivity towards branched aldehyde drops substantially. It is explained by the formation of a more active catalytic species, HRh(CO)2(L) rather than pentacoordinated HRh(CO)2(L)2 18-electron species that might form the R(CO)Rh(CO)2(L)2 acyl intermediate that does not react with hydrogen because of the lack of a vacant coordination site for molecular hydrogen to undergo oxidative addition [21]. It has to be mentioned that the oxidative addition of molecular hydrogen to Rh complexes possessing phosphine and diphosphine ligands is considered as the rate-determining step of hydrogenation. Kinetic studies revealed, however, that the rate-determining step depends on the nature of the ligand and in some cases alkene insertion into Rh-hydride bond becomes the slowest step [22]. The fact that the less hindered (and less basic) ligand decreases the reaction rate, but increases the regioselectivity towards the branched aldehyde, indicates that the regioselectivity is determined in the alkyl–Rh formation step from the π-olefin Rh complex [23].
Due to the great variety of rhodium–ligand systems used in the hydroformylation of styrene, we focused on those catalysts formed from Rh–Cp precursors. By using P-ligand-free (Cp)Rh(CO)2 catalyst, nearly equal amount of linear (10) and branched (9) aldehyde was formed. The application of Rh–Cp complexes containing ester functionalities on the Cp ring slightly increased regioselectivity. Upon addition of PPh3, excellent branched-to-linear aldehyde ratios towards branched aldehydes were, however, obtained. The presence of a phosphine ligand in these rhodium-Cp systems seems to be essential to achieve regioselectivities of practical importance [24].
In those cases, when sterically demanding phosphorus ligands (3, 4, and 6) are used, good to excellent turnover frequencies (TOFs) up to 1000 have been obtained referring to the determining role of coordinatively unsaturated HRh(CO)2(L) species of high activity. It is worth noting that the high activity was accompanied by practically complete chemoselectivities towards aldehydes.
The palladium-catalysed hydroalkoxycarbonylation of styrene (Eq. (2)) resulted in much lower catalytic activities than the rhodium catalysts (containing the same ligands) in hydroformylation. The palladium precursor containing cyclopropanyl ligand 5 formed a more active catalyst than the analogous trans-PdCl2(2)2 complex. In both cases, the formation of the chiral branched ester 12 is highly favoured (Table 1, entries 19 and 20).
| (2) |
3 Experimental
All reactions were carried out under argon using standard Schlenk tube techniques. Toluene was distilled from sodium in the presence of benzophenone. Styrene was freshly distilled before use. The phosphine ligands were prepared by a known deoxygenation reaction of the corresponding precursors bearing P=O moiety by trichlorosilane [25]. The rhodium(III) and Pd(II) complexes used as catalytic precursors were prepared as described earlier [16,17].
3.1 Hydroformylation experiments
In a typical hydroformylation experiment a solution of 0.00125 mmol RhCl2(Cp*)(L) catalyst in 1.5 ml of toluene containing 5 mmol of styrene was transferred into a 20-ml stainless steel autoclave under argon. The reaction vessel was pressurised to 100-bar total pressure with CO/H2 (1:1) and the magnetically stirred mixture was heated in an oil bath. The pressure was monitored throughout the reaction. After cooling and venting of the autoclave, the solution was immediately analysed by gas chromatography.
3.2 Hydroethoxycarbonylation experiments
In a typical experiment, a solution of 0.00125 mmol PdCl2(L)2 catalyst in 1.5 ml of toluene containing 5 mmol of styrene and 0.5 ml of ethanol was transferred into a 20-ml stainless steel autoclave under argon. The reaction vessel was pressurised with CO to 90 bar and the magnetically stirred mixture was heated in an oil bath. The pressure was monitored throughout the reaction. After cooling and venting of the autoclave, the solution was immediately analysed by gas chromatography.
Acknowledgements
K.L. and G.K. thank the OTKA funds (No. T035047 and T 042479, respectively), while I.O. acknowledges the partial support from the Russian Basic Research Foundation (grant No. 02-03-33073).