

1 Introduction
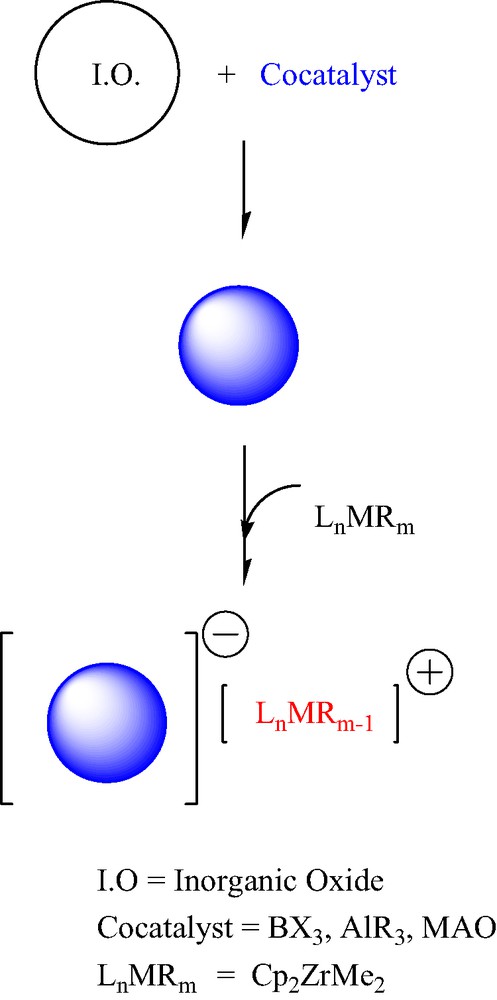
One of the most exciting developments in the areas of catalysis, organometallic chemistry, and polymer science in recent years has been the new polymerisation technologies based on single-site and metallocene coordination olefin polymerisation catalysts [1,2]. The vast number of specifically designed/synthesized transition metal complexes (catalyst precursors) and main-group organometallic compounds (co-catalysts) allows unprecedented control over polymer microstructure, the generation of new polymer architectures, and the development of new polymerisation reactions. A large proportion of commercial polyolefin production is currently achieved by large-scale slurry- and gas-phase polymerisation processes, which require the polymerisation catalysts to be anchored on solid supports. Although supported catalysts are generally less active than homogeneous catalysts, they often offer advantages in producing polymeric products with good morphology and high bulk density. In these supported catalysts, grafted main-group organometallic fragments (co-catalysts) could act as heterogeneous anions on which the metallocenium species are stabilized by ion-pair interactions [2,3] (Scheme 1). Generally, the intimate structure of the active site(s) remains unknown.
Since two decades, the surface organometallic chemistry (SOMC) principle allows to develop access to unique and well-defined organometallic fragments linked to the silica surface [4]. The focus of this paper is to access to a well-defined boron heterogeneous co-catalyst using this approach, in the case of the reaction of tris(pentafluorophenyl)borane, B(C6F5)3 with the silica surface in presence or not of Brønsted bases.
2 Results
Interestingly, although the synthesis of tris(pentafluorophenyl)boron, B(C6F5)3, was known since 1963 [5], very few reports appeared until recently on the reaction of this non-hydrolysable Lewis acid with silica surfaces [6–10]. An attractive feature could be the ability of B(C6F5)3 to enhance the Brønsted acidity of silica by reversible coordination with the surface hydroxyl groups (Eq. (1)) [8]. This behaviour has been observed in solution with Lewis bases such as water [11–16] alcohols [11,16] and silanols [17]. However, the lack of spectroscopic evidences supporting reaction (1) in the solid state justified a more systematic approach, which will be the aim of the first part of this work.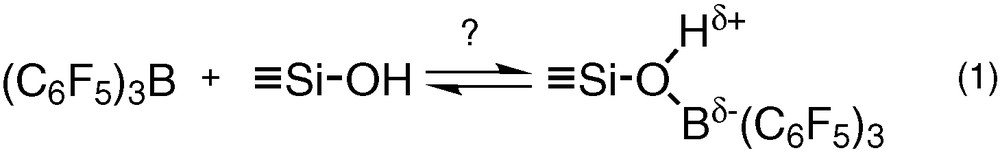
In the second part, the results of the reaction of B(C6F5)3 with silica in the presence of a Brønsted base (C5H5N and Et2NPh) will be reported.
In both cases, the fully characterization by infrared and multinuclear (1H, 13C, 11B) solid-state NMR spectroscopy, elemental analysis, labelling, chemical reactivity, molecular modelling and synthesis of molecular analogues will be described.
2.1 Silica surface
Among the large variety of accessible oxide supports, silica is probably the simplest and the best understood. Its surface is composed of relatively unreactive (at least for moderate temperatures of dehydroxylation) siloxane bridges ≡Si–O–Si≡, and surface hydroxyl groups ≡Si–OH. A huge amount of vibrational spectroscopy and solid-state NMR (1H, 29Si) studies have largely contributed to the determination, the distribution and the reactivity of these surface reactive sites. Some recent reviews are particularly interesting in this field [18–20]. The design of single-site supported organometallic complexes requires the perfect knowledge and the precise control of the surface reactive sites. Their nature and concentration are indeed strongly determining in the final structure of the surface organometallic fragments. A preliminary study of the surface morphology as a function of the pre-treatment temperature is then essential prior to chemical modification.
The support used for our studies was a pyrogenic (Degussa Aerosil© 200). The specific area was 200 m2 g–1 for SiO2–(300,500), 180 m2 g–1 for SiO2–(700,800) and 160 m2 g–1 for SiO2–(1000). The influence of the dehydroxylation temperature on the nature and of the proportion of the reactive surface sites was studied by infrared and 1H NMR spectroscopies.
The dehydroxylation of a compacted silica disk (10 mg) under vacuum was followed by infrared spectroscopy for temperatures ranging from 25 to 1000°C. The overall effect was an increase in the number of isolated silanol groups and an increase in the intensity of the 3747-cm–1 IR band. The consumption of the H-bonded silanols was complete above 500 °C, and only isolated and terminal silanols at 3747 and 3720 cm–1 were still observed at this temperature. For dehydroxylation temperatures above 500 °C, the low wavenumber asymmetry of the residual isolated silanol peak at 3747 cm–1 disappeared and the band became narrower and more symmetrical. For the silica used in our studies, a qualitative approach was carried out by measuring the areas of each type of silanols (bridged and/or isolated) as a function of the dehydroxylation temperatures (Fig. 1). H-bonded silanols represented the majority of the surface hydroxyl groups initially present on silica. As the dehydroxylation temperatures increased, the sum of all silanols (▴) decreased in a same manner than that of bridged silanols (■). Due to the condensation of bridged silanols, the amount of isolated silanols (♦) increased and reached a maximum between 300–400 °C. For higher temperatures (≥ 500 °C), the condensation of isolated silanols occurred and their proportion decreased. At 600 °C, there was no more bridged silanols on the surface. However, since the extinction coefficients of each ν(O–H) vibration have not been calibrated, the relative quantification of the surface silanols by infrared was not possible.
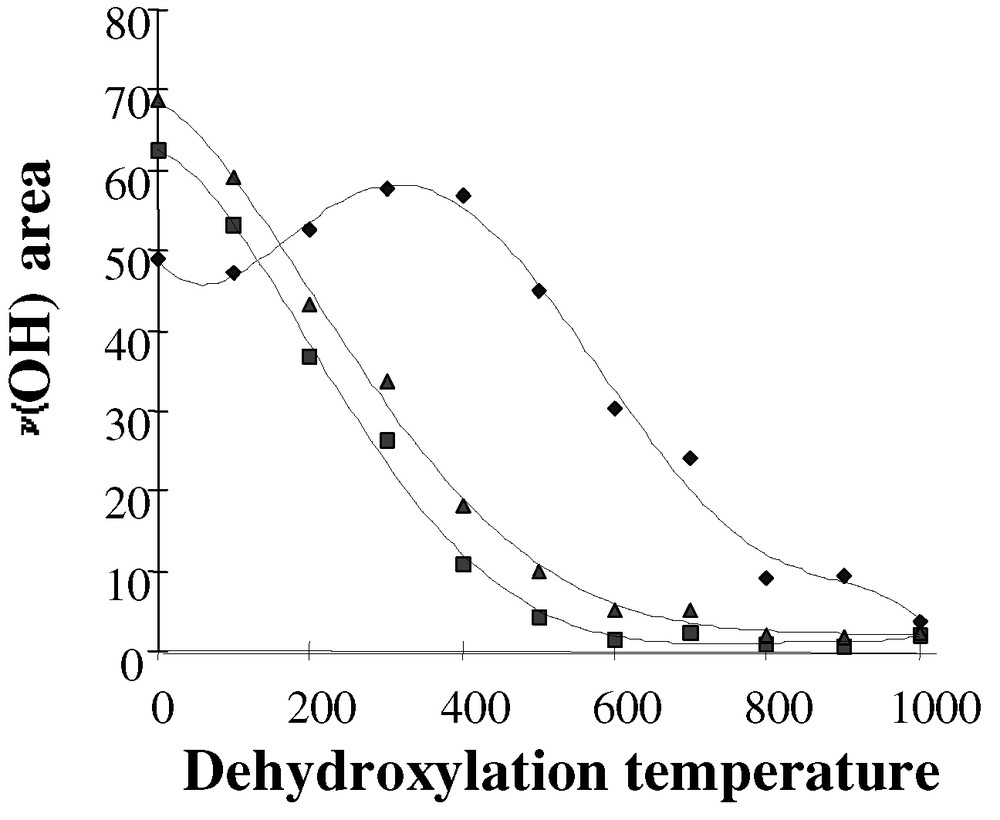
Infrared areas of the ν(OH) vibrations versus dehydroxylation temperatures for (■) bridged silanols, (♦) isolated silanols and (▴) sum of all silanols.
Solid-state 1H NMR spectra of silica samples dehydroxylated at temperatures below 500 °C displayed two peaks at 2.7 and 1.8 ppm. These peaks were respectively assigned to H-bonded, and isolated/geminal surface silanols. Isolated and geminal hydroxyl groups cannot be differentiated by solid-state 1H NMR spectroscopy (the existence of geminal silanols was only evidenced by 29Si NMR spectroscopy [21-23]. For dehydroxylation temperatures above 500 °C, the persistence of the sharp peak at 1.8 ppm demonstrated that only isolated silanols were present on the surface, as shown previously by infrared spectroscopy.
In NMR spectroscopy, the intensity of a signal is directly proportional to the total number of spins present in the sample. Then, relative intensities can be obtained from relative areas of resonance lines, and absolute intensities, from comparison with an external standard. For silica samples, the signals observed for isolated (1.8 ppm) or H-bonded (2.7 ppm) hydroxyls in 1H NMR spectra are therefore proportional to the total amount of surface silanols, this latter being dependant on the mass of the samples and on the pre-treatment temperature. The peaks areas, divided by the mass of the corresponding samples, are representative of the total amount of surface silanols for a given dehydroxylation temperature. Solid-state 1H NMR spectroscopy was then used to quantify the surface silanols for silica dehydroxylated at various temperatures, by reference to NH4Cl as an external standard. The surface concentration αOH of hydroxyl groups (OH/nm2) was then deduced from these data and the surface area as measured by nitrogen adsorption (Table 1).
Experimental data and results for the quantification of surface silanols by solid-state 1H NMR spectroscopy
| Sample | T (°C) | Time (h) a | Weight (mg) | Intensity I b | RGc | Intensity/g (RG = 100)d | δOH mmol g–1e | αOH (OH nm–2)f |
| NH4Cl | — | — | 48.5 | 713 | 64 | 23000 | 74.8 | — |
| SiO2-(150) | 150 | 14 | 19.9 | 112 | 2048 | 275 | 0.894 | 2.69 |
| SiO2-(200) | 200 | 13 | 24.3 | 119 | 2048 | 239 | 0.778 | 2.35 |
| SiO2-(300) | 300 | 14 | 28.0 | 101 | 2048 | 176 | 0.572 | 1.72 |
| SiO2-(350) | 350 | 8 | 27.0 | 162 | 4096 | 146 | 0.476 | 1.43 |
| SiO2-(400) | 400 | 14 | 27.9 | 145 | 4096 | 127 | 0.413 | 1.24 |
| SiO2-(500) | 500 | 10 | 35.2 | 176 | 4096 | 123 | 0.40 | 1.2 |
| SiO2-(800) | 800 | 13 | 35.9 | 100 | 4096 | 68 | 0.223 | 0.6 |
| SiO2-(1000) | 1000 | 14 | 37.0 | 58 | 4096 | 40 | 0.132 | 0.4 |
a Dehydroxylation time at a given temperature.
b Integration on the [(+20)–(–20) ppm] interval.
c Sensitivity factor.
d Intensity/weight reported to a RG value of 100.
e For NH4Cl: calculated on the basis that 1 g of NH4Cl contains 74.8 mmol of hydrogen; for SiO2–(T): (I/g – SiO2-(T)) × (mmolH g–1 – NH4Cl)/(I/g – NH4Cl).
f With specific areas of 200 m2 g–1 for SiO2–(150–500), 180 m2 g–1 for SiO2–(800) and 160 m2 g–1 for SiO2–(1000).
It can be noted that the two analytical methods (1H NMR and TGA) used for Aerosil silica were bulk methods measuring all OH groups, including internal and reactively inaccessible silanol groups. In conclusion, infrared spectroscopy brought qualitative aspects on the evolution and the distribution of surface reactive sites during the condensation process. Solid-state 1H NMR allowed the quantification of all hydroxyl groups present on silica.
2.2 Reactivity of B(C6F5)3 with silica surface
A large excess (5 equiv) of B(C6F5)3 was reacted in toluene with SiO2–(500,700,1000) for 2 h, at room temperature. The resulting pink solids were washed three times with toluene {these washings were collected for further analyses}and dried to give light pink (SiO2–(500,700)) or white (SiO2–(1000)) solids, 1.
The results of elemental analyses revealed a general loading of 0.1–0.2 w% of boron, which corresponds to a concentration of around 0.6 boron per surface silanol. After toluene washings, elemental analyses showed that 30 to 60% of boron has leached. This proved that only weak interactions were involved between B(C6F5)3 and the silica surface (Table 2).
Results of elemental analysis for 1 in function of the dehydroxylation temperature of the silica and the workup
| Runs | T (°C) | OH (nm2) | Before washing | After washing | ||
| % B a | B/OH b | % B a | B/OH b | |||
| 1 | 500 | 1.2 | 0.33 | 0.54 | 0.23 | 0.38 |
| 2 | 700 | 0.7 | 0.24 | 0.70 | 0.10 | 0.29 |
a Weight %.
b Calculated from molar % of B.
IR spectra of solids 1, the bands characteristic of the starting material at 1650, 1523, 1470, 1380 (C6F5 groups[16,24,25] and 1325 cm–1are present (Fig. 2).
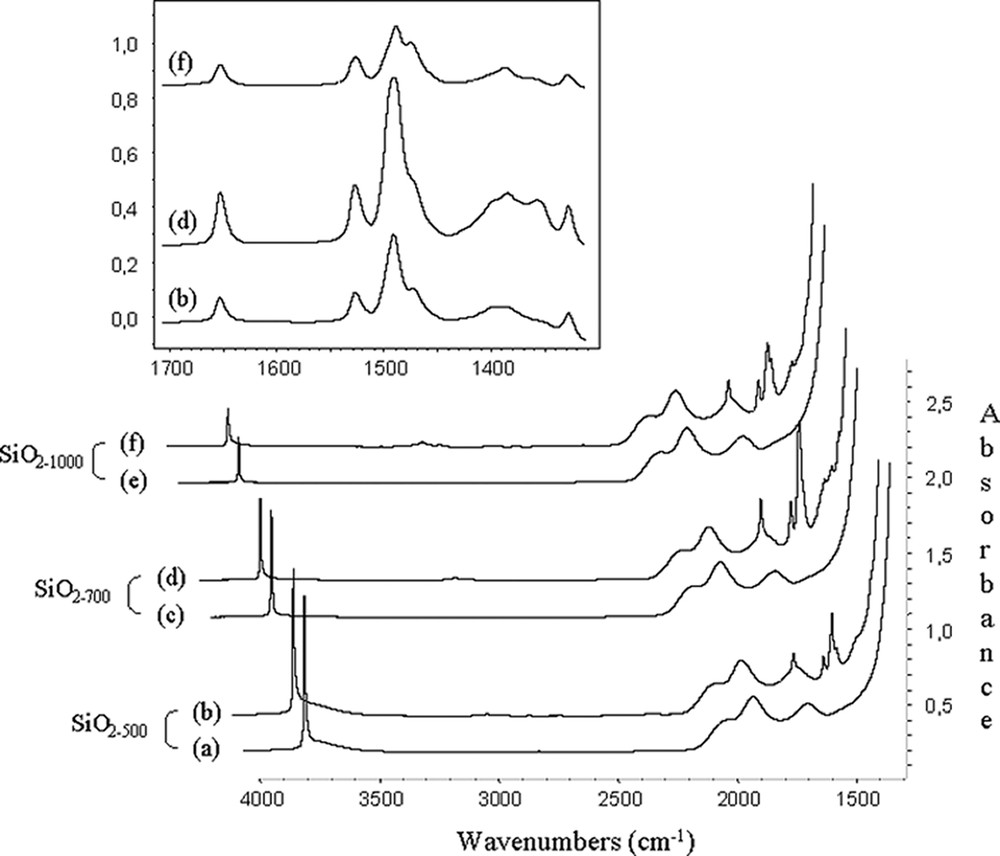
IR spectra of (a) SiO2–(500); (b) SiO2-(500) after reaction with B(C6F5)3 (5 equiv) in toluene; (c) SiO2–(700); (d) SiO2–(700) after reaction with B(C6F5)3 (5 equiv) in toluene; (e) SiO2-(1000); (f) SiO2-(1000) after reaction with B(C6F5)3 (5 equiv) in toluene; and expanded 3800–3200 cm–1 region.
The most significant result was that in these spectra, (Fig. 2), the position and the intensity of the band due to the vibration of the free silanol, ν(≡Si–OH), at 3748 cm–1 was identical before and after reaction with B(C6F5)3. What is more, the 19F NMR spectra of the toluene filtrate (washings) of solids 1 showed only peaks at –129, –142 and –160 ppm, which were unambiguously attributed to ortho-, para- and meta-fluorine atoms of pure B(C6F5)3. No C6F5H was detected by 19F NMR spectroscopy and gas chromatography in the filtrate solution. Likewise, the 1H NMR spectrum of solids 1 revealed no significant difference in chemical shifts of surface isolated silanol groups Si–OH. (Δδ = 0.1 ppm) as expected if the surface Brønsted acidity has been created by interaction of B(C6F5)3 with them [26]. These results indicated that the B–C6F5 bond was not cleaved (Eq. (2)), that there was no interaction of any type between B(C6F5)3 and the oxygen atom of ≡Si–OH (Eq. (3)), and that B(C6F5)3 was only physisorbed onto the silica surface (Eq. (4)).
In order to model the reaction of B(C6F5)3 with silica surfaces, the reactivity of equimolar amounts B(C6F5)3 and R–OH compounds (R = H, Ph3Si, (c-C5H9)7O12Si8) in benzene, at room temperature, was investigated by solution 1H, 13C, 19F and 11B NMR spectroscopy. 19F NMR spectra recorded for these solutions showed a general upfield shift of the para-fluorine after reaction with R–OH (Table 3). Such a shift was characteristic of a change in the boron geometry from trigonal to tetrahedral, induced by coordination with the hydroxy oxygen atom. The difference Δ(δm p) between the chemical shifts of para- and meta-fluorine atoms is strongly connected to the geometry at the boron centre and can be used to determine the strength of coordination of each R–OH compound [27]. The coordination strength at room temperature was shown to increase in the order Ph3SiOH < (c-C5H9)7O12Si8(OH) < H2O (Table 3). It can be noted that no or few HC6F5, resulting from B-C6F5 bond cleavage, was detected in the reaction mixture in our conditions.
1H, 11B and 19F chemical shifts of B(C6F5)3 before and after reaction with R–OH compounds
| Solutions in C6D6 | δ1H (R–OH) | ||||||
| before | after | δ11B | o-C6F5 | p-C6F5 | m-C6F5 | Δ(δm, p) | |
| B(C6F5)3 | — | — | 59 | –128.7 | –141.8 | –160.1 | 18.3 |
| B(C6F5)3 + Ph3SiOH | 3.2 | 4.2 | 40 | –130.4 | –147.9 | –161.0 | 13.1 |
| B(C6F5)3 + (cC5H9)7O12Si8(OH) | 5.2 | 27 | –132.5 | –152.0 | –161.8 | 9.8 | |
| B(C6F5)3 + H2O | 4.6 | 6.8 | –134.8 | –154.2 | –163.0 | 8.8 |
11B NMR spectra recorded after addition of R–OH to B(C6F6)3 at room temperature showed single signals shifted upfield from the initial peak of free B(C6F5)3 (Table 3). 11B chemical shifts are also strongly dependent on the boron geometry and go upfield from trigonal to tetrahedral (neutral then anionic) structures. This confirmed the coordination of B(C6F5)3 on the hydroxylic groups. The upfield shifts observed in 11B NMR spectra of B(C6F5)3 after reaction with R–OH increased in the order Ph3SiOH < (c-C5H9)7O12Si8(OH) < H2O (Table 3) and corroborated the coordination strengths evidenced by 19F NMR spectroscopy.
We can then conclude from these studies that at the opposite of silanol groups, the molecular compounds R–OH [ROH = Ph3SiOH, (c-C5H9)7O12Si8(OH)H2O] form a stable complex with B(C6F5)3 (Eq. (5)).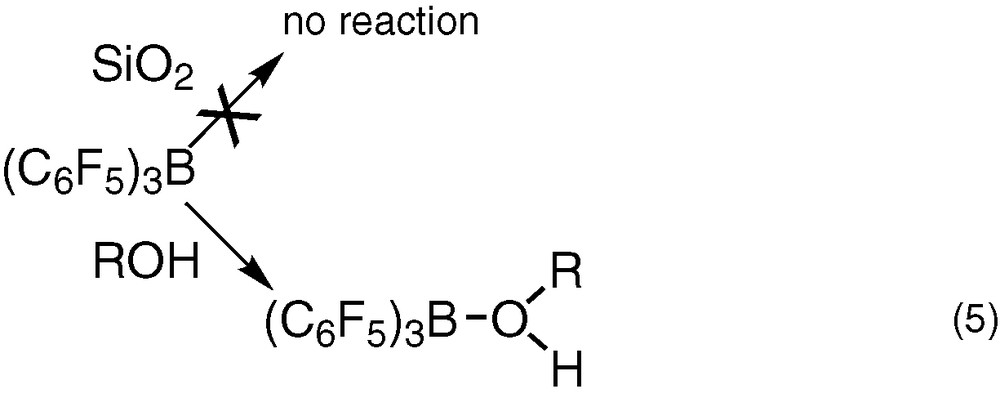
2.3 Reactivity of B(C6F5)3 with silica surface in presence of a Brønsted base
The above results demonstrated that there was no interaction between B(C6F5)3 and ≡Si–OH. At the opposite, pyridine forms hydrogen bonds with ≡Si–OH [28-30] (Eq. (6)). This hydrogen bond induces a higher negative charge on oxygen atom of ≡Si–OH, which could favour the formation of ion pair [≡Si–O B(C6F5)3]–[HNC5H5]+. To verify this hypothesis, the reaction of toluene solution of C5H5N and B(C6F5)3 (1:1) with SiO2 was performed.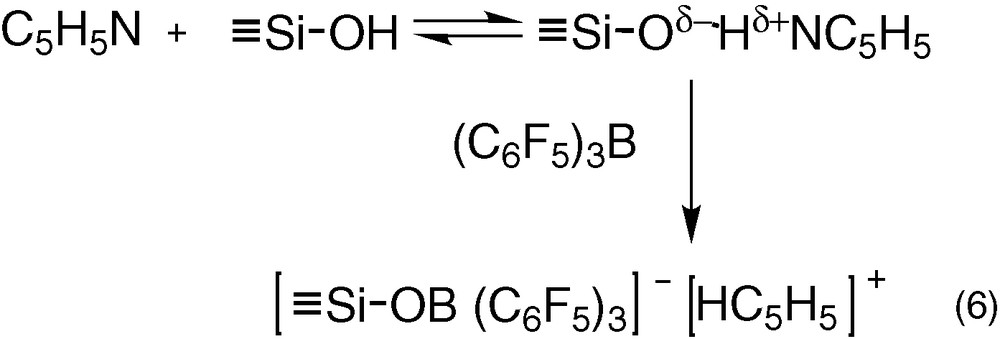
2.3.1 Reactivity of 1 with pyridine
Pink silica pellets of 1 prepared by impregnation of B(C6F5)3 on SiO2–(500,700,1000), were contacted with a vapour pressure of pyridine under static vacuum at room temperature. The ν(≡Si–OH) band at 3747 cm–1 due to free silanol disappeared and simultaneously a broad band resulting from hydrogen bonding between the surface silanols and pyridine appeared near 2900 cm–1. Aromatic ν(C–H) and ν(C=C) vibrations were also observed at 3081, 3025, 3002 cm–1 and in the [1650–1500 cm–1] region respectively, and were characteristic of the pyridine. The bands initially present at 1648 and 1523 cm–1 in 1 for the C6F5 groups were unaffected pyridine (respectively 1645 and 1519 cm–1 after desorption), while the strong one at 1483–1473 cm–1 was shifted by about 20 cm–1 to lower wavenumbers (1465 cm–1). Other vibrations observed in 1 at 1394, 1383 and 1323 cm–1 disappear almost entirely after contact with pyridine. The same tendencies were noticed for the molecular compounds B(C6F5)3 and (C6F5)3B.NC5H5 adduct. The excess of pyridine was removed under vacuum (10–5 mbar) at room temperature. In the infrared spectra, the position of the peaks at 1632, 1492 and 1447 cm–1 indicates the presence of pyridine bonded to a Lewis acid site [28–30]. The shift of the peak of pyridine 8a mode [30] from 1583 to 1631 cm–1 (Δν = 43 cm–1) suggests the presence of strong strength Lewis acid. On the contrary, no bands at 1638, 1620 and 1540 cm–1, characteristic of pyridine bonded to a Brønsted acid site on 1, are observed (Fig. 3).
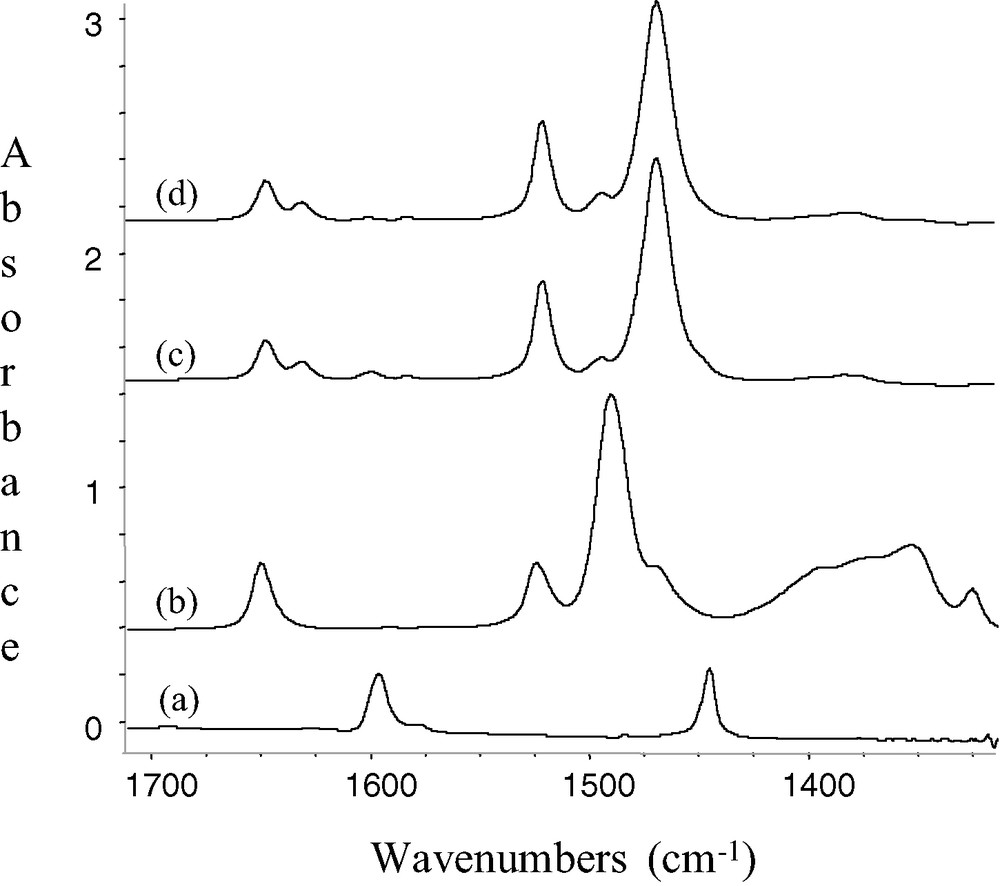
IR spectra(silica spectra subtracted) of (a) SiO2-(700) + C5H5N; (b) SiO2-(700) after reaction with B(C6F5)3 (5 equiv) in toluene; 1 (c) 1 + C5H5N (d) (C6F5)3B.NC5H5/KBr; and expanded 1700–1400 cm–1 region.
Solid-state 1H NMR spectrum of 1 after reaction with C5H515N shows a broad peak at 2.3 ppm, attributed to hydrogen-bound silanol groups, and three peaks at 7.4, 7.8 and 8.6 ppm characteristic of meta, para and ortho protons of the heterocycle. The solid state 15N NMR spectra displayed a resonance at –150 ppm, characteristic of Lewis acidic site [26,30–34], unambiguously assigned to the Lewis acid-base adduct (C6F5)3B–NC5H5, as demonstrated by comparison with the 15N NMR spectrum of (C6F5)3B–(15N C5H5) in the solid state (δ15N = –159.2 ppm). As by infrared studies, no Brønsted site acid was evidenced onto B(C6F5)3-modified silica 1 by solid-state 1H and 15N NMR spectroscopies. The strong Lewis acidity detected on 1 is only due to homogeneous (C6F5)3B-(15N C5H5) physisorbed on the surface. The reaction of B(C6F5)3 and C5H5N (1:1) in toluene afforded quantitatively the complex (C6F5)3B.N C5H5, 2, which did not react with SiO2, It was only physisorbed (Eq.(7)).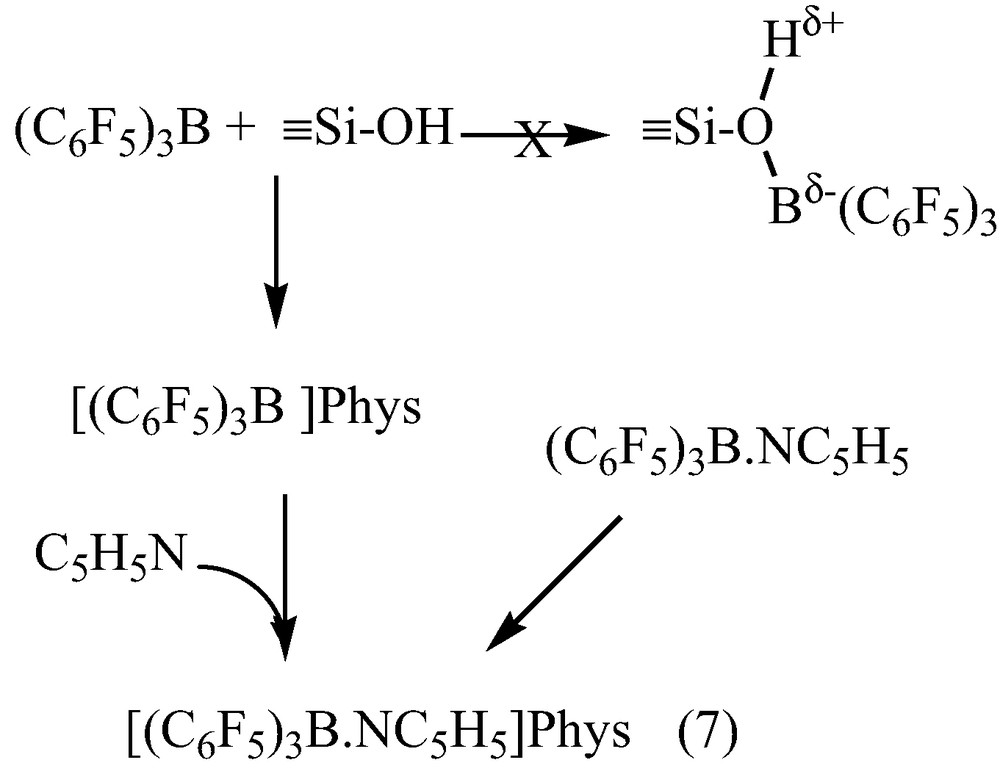
2.3.2 Reactivity of 1 with diethylaniline
The addition of one equivalent of NEt2Ph to B(C6F5)3 in aromatic solvents led instantaneously to a pink coloration of the solution, S. The stoichiometric reaction of B(C6F5)3 and NEt2Ph at room temperature, in an aromatic solvent, has been investigated by 1D and 2D NMR of 1H,13C, 11B, 19F and 15N [35]. No adduct Et2PhN.B(C6F5)3 was formed. An equilibrium has been evidenced between free (C6F5)3B (40%) and Et2NPh, imminium salt (HB(C6F5)3)–(HNEt2Ph)+ (30% ), and zwitterionic stereoisomers E and Z-[PhEtN+=CH–CH2B–(C6F5)3] in a 3:2 ratio (30%) [36]. In the presence of a protic compound, this equilibrium is totally displaced towards the ionic form [RO–B(C6F5)3]–[HNEt2Ph]+ [36].
Silica pellets 2 were prepared by impregnation of S on SiO2–(300,500,800). The IR band ascribed to isolated silanols at 3747 cm–1 disappeared totally and was replaced by broad bands at 3681, 3624 cm–1 (ν(OH) of interacting silanols) and by a sharp band at 3232 cm–1. The intensity of the bands at 3681, 3624 cm–1 decreases inversely with the dehydroxylation temperature; simultaneously, that of the band at 3232 cm–1 increases (Fig. 4).
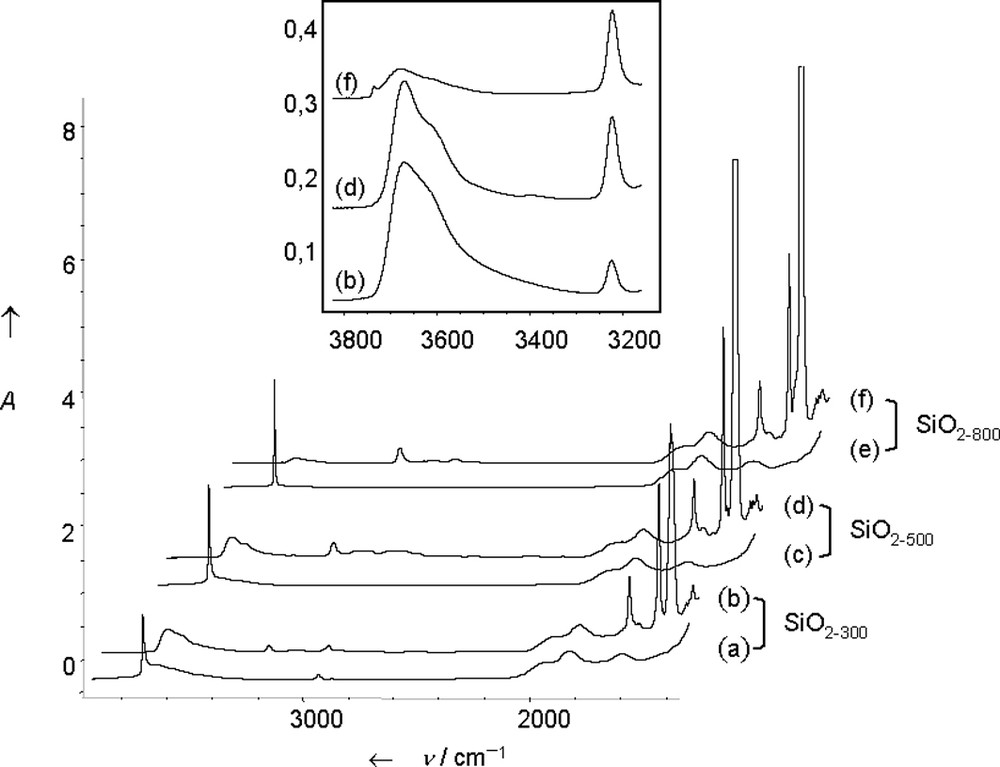
IR spectra of (a) SiO2–(300); (b) SiO2–(300) after reaction with S (S = B(C6F5)3 + NEt2Ph, 1:1 mixture, 5 equivalents); (c) SiO2–(500); (d) SiO2–(500) after reaction with S; (e) SiO2–(800); (f) SiO2–(800) after reaction with S; and expended 3800–3200 cm–1 region.
The use of labelled 2H or 18O silica proved that the band at 3232 cm–1 was due to a ν(N–H) vibration [36]. The structure of the supported species was confirmed by solid-state 1H NMR [δC6H5 = 7.2, δCH2 = 3.4, δCH3 = 1 ppm], solid-state CP-MAS 13C NMR [δCH3 = 39, δCH2 = 51 ppm] and solid-state 11B NMR [δ = –8 ppm], in agreement with the 11B chemical shift (solid state) of molecular analogous (–6.9 and –7 ppm), or literature data [6]. It is characteristic of an anionic borate fragment, i.e. [≡SiO–B(C6F5)3]–, in which the boron atom is tetracoordinated.
Elemental analyses of 2 /SiO2–(300,500,700,800) (Table 4) indicate a slight decrease in the weight percentage of boron, from 0.23 to 0.17%, when the dehydroxylation temperature of the silica is increasing from 300 to 800 °C. But no leaching of boron occurred after washing(see experimental part).
Elemental analysis of 2 in function of the dehydroxylation temperature of SiO2
| T (°C) | OH (nm2) | %B a | %N a | B/N b | F/B b,c | C/(B+N) b | B/OHd | N/OH d |
| 300 | 1.7 | 0.23 | 0.30 | 0.99 | 10.6 | 12.1 | 0.38 | 0.38 |
| 500 | 1.2 | 0.21 | 0.27 | 1.01 | 13.6 | 14.3 | 0.49 | 0.48 |
| 500 | 1.2 | 0.22 | 0.29 | 1 | 12 | 13 | 0.51 | 0.52 |
| 700 | 0.7 | 0.16 | 0.18 | 1.15 | 12.3 | 15.6 | 0.71 | 0.61 |
| 800 | 0.6 | 0.16 | 0.21 | 0.99 | 16.2 | 16.4 | 0.82 | 0.84 |
| 800 | 0.6 | 0.17 | 0.24 | 0.92 | 12.1 | 15.2 | 0.88 | 0.96 |
| Theory | 1 | 15 | 14 |
a Weight %.
b Molar ratios.
c Titration values obtained by conductimetry for fluorine in presence of boron were lowered by formation of BF4– anion.
d Calculated from respective weight percentages of boron and nitrogen.
As demonstrated by infrared spectroscopy and elemental analysis, it must be emphasized that silanol groups could be only partially converted to ionic sites, depending on dehydroxylation temperatures. Indeed, weight percentages of boron corresponded to a proportion of modified silanols ranging from 37 to 96% when the pre-treatment temperatures of the silica increased from 300 to 800 °C.
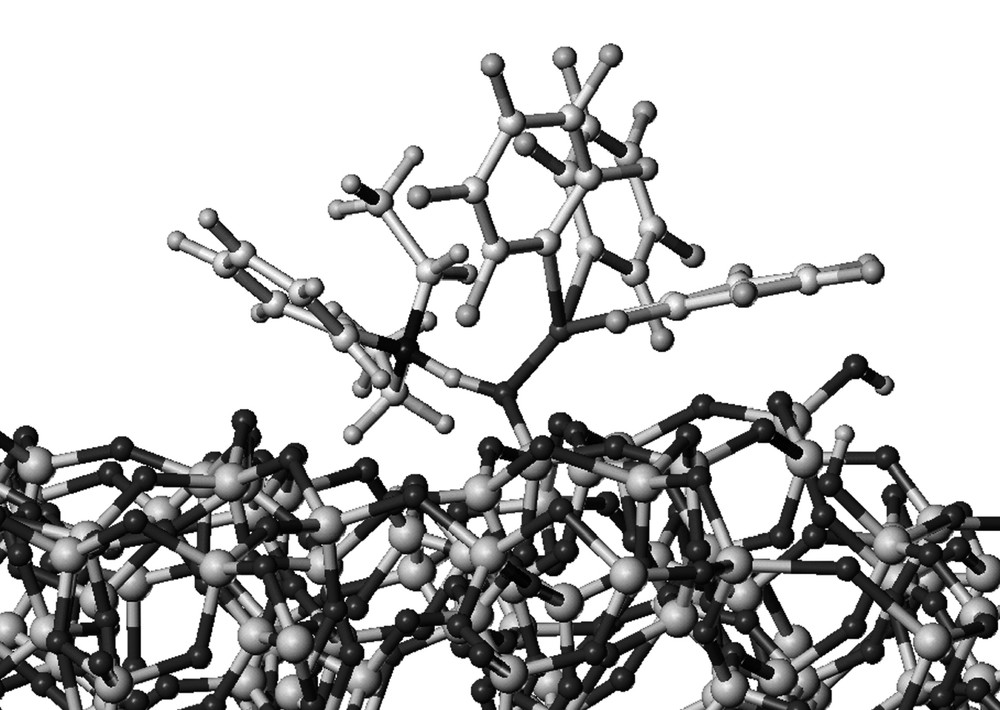
In order to check if the experimental weight percentage of boron corresponded to the highest loading achievable on the surface, modelling of 1 was performed using Sybyl computer modelling program. This entity was then grafted on a model of a partially dehydroxylated silica particle SiO2–(500) and minimized using the molecular mechanics Tripos force field [37–38]. A three-dimensional representation of the grafted model is depicted in (Fig. 5). The theoretical value obtained for the length of the B–O bond (1.567 Å) in the modelled complex was in the range of those reported for related silsesquioxane compounds (1.505–1.495 Å) [16,24–25]. The projected area of such a complex on the silica particle was estimated to 1.27 nm2. According to the silica specific area (200 m2 g–1), this projection corresponds to a theoretical value of 0.24 w% of boron on a saturated surface. This result is in good agreement with the experimental percentages obtained on SiO2–(500) (0.21–0.22 w%).
Al spectroscopic and analytic data, as well as the similarities with those of molecular compounds [HNEt2Ph]+[(C6F5)3BOR]– (R = H, SiPh3, Si8O12(c-C5H9)7) [17,36,39] confirmed the already proposed [6,7,9] structure for 2 (Eq. (8)).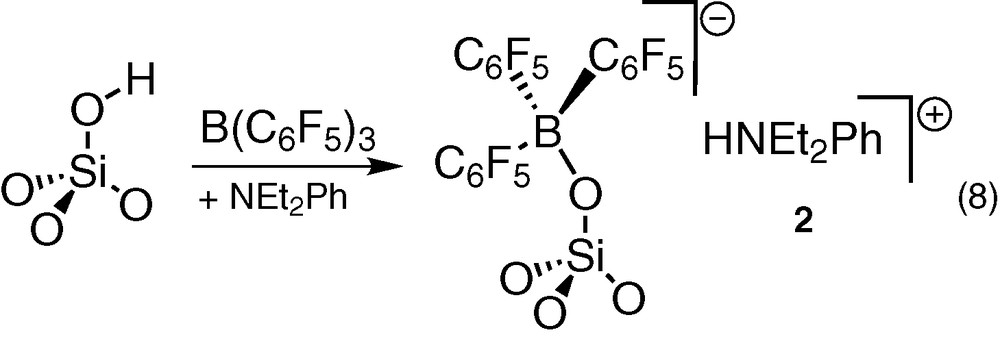
3 Conclusion
Infrared and solid-state 1H NMR spectroscopies have shown that the dehydroxylation temperature can be used to understand and control the surface morphology of silica, e.g. the nature, distribution and concentration of reactive sites. It was shown that uniformity in surface reactive sites was only found for highly dehydroxylated silica surfaces (600–800 °C) as isolated silanols. At lower dehydroxylation temperatures (T ≤ 400 °C), these latter coexist with H-bonded silanols. Moreover, the precise quantification of hydroxyl groups on silica allowed us to control subsequently the stoichiometry of surface reactions involved in the grafting of organometallic complexes.
Regarding the reaction of B(C6F5)3 with silica surfaces, the coordination of the boron centre to a surface silanol group was not proved in the solid state. On the contrary, infrared and solid-state 1H and 11B NMR studies showed that the silanol groups were not affected by the presence of B(C6F5)3 on silica surface, and that the boron atom has kept a trigonal planar configuration. As no or few HC6F5 was detected in the impregnation solutions, the cleavage of B–C bounds by surface silanols was ruled out as a major reaction pathway. B(C6F5)3 was only physisorbed onto the silica surface.
In the presence of the strongly nucleophilic pyridine, the formation of the adsorbed Lewis acid-base adduct, (C6F5)3B.NC5H5, was privileged, displaced the reaction (6) totally towards the right. Such a reaction was also described for homogeneous silsesquioxane models.
The interaction of B(C6F5)3 with silica in the presence of Et2NPh yielded to [≡Si–O–B(C6F5)3]–[HNEt2Ph]+. The formation of [≡Si–O–B(C6F5)3]–[HNEt2Ph]+is probably due to the nature of NEt2Ph which cannot, due to the steric congestion (B-strain), form a complex with B(C6F5)3, at the opposite of pyridine and Me2NPh [36]. The use of the stoichiometric nature of surface organometallic chemistry allowed us to transform selectively each surface silanol group [≡SiOH] into a unique and well-defined ionic entity [≡SiO–B(C6F5)3]–[HNEt2Ph]+ only when the pre-treatment temperature of the silica was 800 °C and above. Out of these conditions, on the silica surface, two types of surface sites were found: the free silanol groups [≡SiOH] and the ionic [≡SiO–B(C6F5)3]–[HNEt2Ph]+ groups in relative proportion of 3:2 for SiO2–(300) and 1:1 for SiO2–(500). This surface [≡SiO–B(C6F5)3]–[HNEt2Ph]+ is the first example of the selective preparation and full characterization of a heterogeneous non-coordinating anion/free cation system. This surface fragment can be used as a very promising building block to generate cationic heterogeneous anions on which the metallocenium species are stabilized by ion-pair interactions. Accordingly, the reaction of Cp*ZrMe3 with [≡SiO–B(C6F5)3]–[HNEt2Ph]+, afforded, by an irreversible process of methane elimination, to an unique and well-defined cationic silica-supported metallocenium species [≡SiO–B(C6F5)3]–[Cp*ZrMe2(NEt2Ph)]+, which was an active olefin polymerisation catalyst [41].
4 Experimental
4.1 General procedures
Solid-state NMR spectra were recorded on a Bruker DSX-300 spectrometer equipped with a standard 4-mm double-bearing probe head and operating at 75.47, 96.31, 30.43, and 300.18 MHz for 13C, 11B, 15N and 1H, respectively. Chemical shifts were given with respect to external references (1H, 13C, TMS at 0 ppm; 11B, BF3.OEt2 at 0 ppm; 15N, CH3NO2 at 0 ppm). The samples were introduced in the zirconia rotor in a glovebox and tightly closed. Solution NMR spectra were recorded on Bruker AC 200 MHz (19F), AC 300 MHz (1H, 13C), DRX 300 MHz (11B) and DRX 500 MHz (1H, 13C, 11B, 15N) spectrometers. Chemical shifts were reported in ppm and referenced to residual solvent resonances (C6D6: 7.15 for 1H, 128 for 13C; CD2Cl2: 5.32 for 1H, 53.8 for 13C), or external standards (19F, CFCl3 at 0; 11B, BF3.OEt2 at 0; 15N, CH3NO2 at 0). Infrared spectra were recorded under vacuum on a Nicolet 550 FT spectrometer by using an infrared cell equipped with CaF2 windows, allowing in situ studies. Typically, 16 scans were accumulated for each spectrum. KBR pellets were prepared for molecular compounds. Elemental analyses were performed by the Central Analysis Service of the CNRS at Solaize.
All operations were performed in the strict absence of oxygen and water under a purified argon atmosphere using glovebox (Jacomex or MBraun) or vacuum-line techniques. Toluene and pentane were distilled under argon from Na/K alloy, degassed and stored under argon over Na (toluene) or 3–Å molecular sieves (pentane). C6D6 (SDS – 99.6%) and CD2Cl2 (SDS – 99.6%) were degassed by three cycles of ‘freeze-pump-thaw’ and dried over freshly regenerated 3 Å molecular sieves. Pyridine (Aldrich, 99%) was dried on solid KOH and distilled under argon, degassed and stored over 3–Å molecular sieves. Labelled 15N pyridine (Aldrich, 99%) was degassed and store over 3Å molecular sieves. B(C6F5)3 (Merck Chemicals, > 97%) was dried on Me3SiCl [40], purified by vacuum sublimation and its purity was checked by 19F NMR before using. The 3,5,7,9,11,13,15-heptacyclopentyl-pentacyclooctasiloxan-1-ol and the triphenylsilanol were purchased from Aldrich Chemical and dried under vacuum before use. The synthesis of B(C6F5)3.OH2 [13] [16], B(C6F5)3.N C5H5 [17], and [Z–O B(C6F5)3]–[HNEt2Ph]+ with Z–OH, Z = H, Ph3Si, (c-C5H9)7O12Si8 [36], or silica were performed according to literature procedures. Silica support (Aerosil® Degussa, 200 m2 g–1) was compacted to a disk (30 mg) for infrared studies or was hydrated, dried (80 °C) and crushed to prepare large quantities (1–2 g) for NMR studies and elemental analyses. Before reaction, to prepare large quantities, silica was calcinated at 400 °C in air for 4 h, and dehydroxylated at the desired temperature (500, 700, 800 or 1000 °C) under high vacuum (10–5 Torr) for 12 h (referred respectively as SiO2–(500), SiO2–(700) and SiO2–(1000)), except in the case of silica dehydroxylated at 300 °C (referred as SiO2–(300)), which was calcinated at 400°C in air for 4 h, rehydrated with distilled water and then dehydroxylated at 300 °C for 12 h. The surface areas were measured by N2 adsorption and treatment of data using BET method.
4.2 Preparation of the B(C6F5)3-modified silica supports, 1, and reactivity with pyridine
4.2.1 For infrared experiments
A disk of compacted SiO2–(700) (30 mg) was reacted under argon and at room temperature with a solution of 16 mg (0.031 mmol, 5 equiv) of B(C6F5)3 in 10 ml of toluene during 2–3 h. The pink pellet was then washed with three fractions of 10 ml of dry toluene and dried 1 h at room temperature under 10–5 mbar. IR spectra were recorded before and after washings. IR (cm–1): 3748 (s) ν(O–H)isolated, 1650 (m) C6F5, 1523 (m) C6F5, 1491 (s) C6F5, 1470 (m) C6F5, 1395 (m), 1380 (m) C6F5, 1354 (m), 1325 (m). The same procedure was used for SiO2–(500) and SiO2–(1000).
4.2.2 For large-scale preparation (NMR and elemental analysis samples)
A solution of 107 mg (0.21 mmol–1 equiv) of B(C6F5)3 in 10 ml of dry toluene was filtered under argon onto 1 g of compacted SiO2–(700). The pink suspension was stirred under argon for 1–2 h at room temperature. The colourless impregnation solution was then either evaporated to give 1e, or filtered, removed with a syringe and the solid washed three times with 10 ml of dry toluene to give 1w. The pink solids 1e and 1w were finally dried under vacuum (10–5 mbar) for 4 h at room temperature. Elemental analysis: w% B = 0.13. 1e, 1w: solid-state 1H NMR (δ): 2.1 (s, isolated Si–OH). No peak detected in solid-state 11B NMR spectra. The same procedures were used for SiO2–(300) and SiO2–(500) and similar results were obtained.
4.3 Adsorption of pyridine on 1
4.3.1 For infrared experiments
A vapour pressure of pyridine was condensed at 73 K onto B(C6F5)3-impregnated silica pellets 1 (30 mg) prepared as described above. After warming to room temperature, the colour of the solid changed from pink to white. The excess of pyridine was then desorbed under vacuum (10–5 mbar) during 1 h at room temperature. Infrared spectra were recorded at various desorption times and at room temperature. IR (cm–1): 3747 (s) ν(O–H)isolated, 1646 (m) C6F5, 1631 (m) L-bonded C5H5N, 1599 (m) H-bonded C5H5N, 1583 (w) physisorbed C5H5N, 1520 (m) C6F5, 1492 (w) L+H-bonded C5H5N, 1469 (s) C6F5, 1446 (w) L+H-bonded C5H5N, 1396 (w) C6F5, 1384 (w) C6F5.
4.3.2 For larger scale preparation (NMR and elemental analysis samples)
Same procedure with a vapour pressure of labelled 15N pyridine onto 100 mg of B(C6F5)3-impregnated silica 1e and 1w. After reaction with pyridine (15N), the colour of the solids changed from pink to white at room temperature. 1w: Solid-state 1H NMR (δ): 2.2 (br.s, H-bonded Si–OH), 7.4 (s, m-C5H5N), 7.8 (s, p-C5H5N), 8.5 (s, o-C5H5N). Solid-state 15N NMR (δ): –106 (s, Lewis acidic centre), ε –153 (s, (C6F5)3BNC5H5). No peak observed in 11B NMR spectra. 1e: solid-state 1H NMR (δ): 8.2 (br.s, C5H5N), 1.9 (s, ≡SiOH). Solid-state 15N NMR (δ): –102 (s, Lewis acidic centre), –160 (s, C6F5)3BNC5H5). Solid-state 11B NMR (δ): –7.6 (s, C6F5)3B.NC5H5), –11.8 (s).
4.4 Reactivity of B(C6F5)3 with H2O
Addition of 7 μl (0.39 mmol) of distilled water to a solution of 206 mg (0.4 mmol, 1 equiv) of B(C6F5)3 in 12 ml of pentane led to precipitation of a white solid. The suspension was stirred overnight, and the crude product obtained after filtration was washed three times with pentane (4 ml) and dried 1 h under vacuum. Yield (175 mg, 82%) 1H NMR (C6D6, δ): 4.6 (s, OH). 13C NMR (C6D6, δ): 148.0 (d, 1JC–F = 242 Hz, o-C6F5), 141.5 (d, 1JC–F = 254 Hz, p-C6F5), 137.6 (d, 1JC–F = 255 Hz, m-C6F5), 114.6 (br.s, i-C6F5). 19F NMR (C6D6, δ): –134.8 (d, 3JF–F = 23 Hz, 6F, o-C6F5), –154.3 (t, 3JF–F = 21 Hz, 3F, m-C6F5), –162.5 (t, 3JF–F = 23 Hz, 6F, m-C6F5). 11B NMR (C6D6, δ): 6.8 (s, (C6F5)3B–OH2). IR (KBr, cm–1): 3565 (m), 3502 (m), 1652 (m), 1610 (w), 1523 (s), 1473 (s), 1382 (m), 1292 (m), 1270 (w), 1246 (w), 1122 (m), 1112 (m), 1093 (m), 1014 (w), 974 (s).
4.5 Reactivity of B(C6F5)3 with Ph3SiOH
In a NMR tube, 75 mg (0.15 mmol – 1 equiv) of B(C6F5)3 was added at room temperature to a solution of 40 mg (0.14 mmol) of Ph3SiOH in 0.5 ml of d6-benzene. A clear colourless solution was obtained after stirring. 1H NMR (C6D6, δ): 7.5 (d, 3JH–H = 7.5 Hz, 6H, o-C6H5), 7.1 (m, 9H, p- and m-C6H5), 4.2 (br.s, 1H, –OH). 13C NMR (C6D6, δ): 148.2 (d, 1JC–F = 244 Hz, o-C6F5), 137.7 (d, 1JC–F = 255 Hz, m-C6F5), 135.6 (s, o-C6H5), 133.1 (s, i-C6H5), 131.0 (s, p-C6H5), 128.4 (s, m-C6H5), p- and i-C6F5 carbons non resolved. 19F NMR (C6D6, δ): –130.4 (d, 3JF–F = 17 Hz, 6F, o-C6F5), –147.9 (br.s, 3F, p-C6F5), –161.0 (m, 6F, m-C6F5). 11B NMR (C6D6, δ): 40 (br.s, Ph3Si(OH)B(C6F5)3).
4.6 Reactivity of B(C6F5)3 with (c-C5H9)7O12Si8(OH)
In a NMR tube, 75 mg (0.15 mmol, 1 equiv) of B(C6F5)3 was added at room temperature to a solution of 135 mg (0.15 mmol) of (c-C5H9)7O12Si8(OH) in 0.5 ml of d6-benzene. A clear colourless solution was obtained after stirring. 1H NMR (C6D6, δ): 5.2 (br.s, 1H, OH), 1.8 (m, 14H, CH2 C5H9), 1.6 (m, 28H, CH2 C5H9), 1.4 (m, 14H, CH2 C5H9), 1.1 (m, 7H, CH C5H9). 13C {1H} NMR (C6D6, δ): 148.4 (d, 1JC–F = 240 Hz, o-C6F5), 141.9 (d, 1JC–F = 254 Hz, p-C6F5), 137.6 (d, 1JC–F = 253 Hz, m-C6F5), 27.7, 27.6, 27.5, 27.4, 27.3 (s, CH2 C5H9), 22.5, 22.3, 21.9 (s, CH C5H9). 19F NMR (C6D6, δ): –132.6 (br.s, 6H, o-C6F5), –152.0 (br.s, 3H, p-C6F5), –161.8 (m, 6H, m-C6F5). 11B NMR (C6D6, δ): 27 (br.s, (c-C5H9)7O12Si8(OH)B(C6F5)3).
4.7 Preparation of (C6F5)3B–NC5H5
To a solution of 0.248 g (0.48 mmol) of B(C6F5)3 in 5 ml of toluene was added 0.042 ml (0.52 mmol) of pyridine to give a colourless solution. After stirring for 1 h, the toluene was evaporated under vacuum to leave a white solid which was washed twice with pentane (2 ml) and dried for 1 h at room temperature. Yield = 0.234 g (0.40 mmol – 82%). 1H NMR (C6D6, δ): 7.94 (d, 3JH–H = 5 Hz, 2H, o-C5H5N), 6.57 (t, 3JH–H = 7 Hz, 1H, p-C5H5N), 6.24 (t, 3JH–H = 7 Hz, 2H, m-C5H5N). 13C {1H} NMR (C6D6, δ): 148.3 (d, 1JC–F = 243 Hz, o-C6F5), 146.4 (s, o-C5H5N), 141.7 (s, p-C5H5N), 140.8 (d, 1JC–F = 232 Hz, p-C6F5), 137.7 (d, 1JC–F = 242 Hz, m-C6F5), 124.9 (s, o-C5H5N), 118.6 (br.s, i-C6F5). 19F NMR (C6D6, δ): –131.4 (d, 3JF–F = 21 Hz, 6F, o-C6F5), –155.4 (t, 3JF–F = 21 Hz, 3F, p-C6F5), –162.6 (t, 3JF–F = 19 Hz, 6F, m-C6F5). 11B NMR (C6D6, δ): –3.7 (s, (C6F5)3B–NC5H5). 15N NMR (C6D6, δ): –158.2 (s, (C6F5)3B–NC5H5). IR (KBr, cm–1): 1646 (m) C6F5, 1631 (m) L-bonded C5H5N, 1578 (w) C5H5N, 1520 (s) C6F5, 1495 (m) L-bonded C5H5N, 1462 (vs) C6F5, 1385 (m) C6F5, 1378 (m) C6F5, 1293 (m), 1283 (m), 1252 (m), 1242 (m), 1224 (m), 1162 (w), 1115 (m), 1098 (m), 1028 (w), 991 (s) C6F5, 981 (s) C6F5, 962 (m) C6F5.