

1 Introduction
La RMN quantitative du deutérium permet de déterminer aujourd'hui en routine la distribution isotopique naturelle pour chaque site d'une molécule [1]. Cette méthode, brevetée sous le nom de SNIF–NMR™ (Site-Specific Natural Isotope Fractionation by Nuclear Magnetic Resonance, Trade Mark d'Eurofins, Nantes, France), permet de mesurer les variations significatives de cette distribution isotopique en fonction du mode de formation de la molécule [2]. Le champ d'application de cette méthode comprend notamment la détection de la chaptalisation des vins [3,4], l'authentification de l'origine des produits [5–11] et la détermination des mécanismes de biosynthèse [12,13]. Dans le cas du 2H, très peu abondant, l'abondance isotopique peut être assimilée au rapport isotopique ou (D/H).
La surface (Si) sous une raie d'un spectre RMN est directement proportionnelle au nombre (N) de noyaux qui produisent cette raie. N est proportionnel à la concentration c de l'isotopomère comportant le noyau étudié à la position i.
| (1) |
Lorsque l'abondance isotopique moyenne sur l'ensemble des isotopomères, , peut être mesurée (par exemple par SMRI), on peut écrire les équations :
| (2) |
| (3) |
Lorsqu'il est possible d'introduire une référence interne dans l'échantillon, il n'est plus nécessaire de connaître . Cette référence interne doit être soigneusement choisie et mise en œuvre. L'abondance isotopique et la pureté devant être parfaitement connues, soit une calibration préalable est nécessaire, soit le composé est un standard commercial calibré, qui induit un surcoût significatif. Le composé doit présenter une co-solubilité avec l'échantillon, un (des) déplacement(s) chimique(s) différent(s) et une bonne stabilité chimique en présence de l'échantillon. Afin de limiter la quantité de référence à ajouter, le nombre de noyaux équivalents doit être élevé. Enfin, pour ne pas induire une augmentation du temps d'expérience, le T1 de la référence doit être inférieur ou égal à ceux du composé étudié. Dans le cas de l'analyse de l'éthanol, la référence officielle est la tétraméthylurée (TMU).
La technique de référence ERETIC (Electronic REference To access In vivo Concentration) a été conçue afin d'éviter l'ajout, contraignant, voire impossible, d'une substance de référence en RMN in vivo [14,15]. Elle a ensuite été appliquée à la mesure de concentrations en RMN–1H [16–18]. Cette étude propose d'éliminer les contraintes de la référence interne en utilisant la méthode ERETIC pour la mesure des abondances isotopiques 2H.
L'abondance isotopique spécifique a ainsi été mesurée parallèlement grâce aux deux méthodes (référence interne et ERETIC), sur quatre éthanols d'origines (et donc d'abondances isotopiques spécifiques) différentes. Un cinquième éthanol a été utilisé comme référence.
2 Matériels et méthodes
2.1 Échantillons analysés
Les cinq éthanols utilisés proviennent d'origines botaniques différentes : raisin, betterave, canne, blé et maïs. Les échantillons ont été obtenus par fermentation, puis distillation, afin qu'ils contiennent plus de 95% d'éthanol, et conservés sur tamis moléculaire.
Une première série d'échantillons est préparée dans des proportions standards (3.3 ml d'éthanol pour 1.8 ml de TMU, 150 μl de substance-lock C6F6) : c'est la série no 1. La TMU employée est fournie par Eurofins, Nantes. Son abondance isotopique a été calibrée selon un protocole officiel européen à 84,44 ppm [19].
Une deuxième série de tubes est préparée à partir des mêmes échantillons, simultanément. Ces tubes ne contiennent alors quasiment que de l'éthanol (5,1 ml d'éthanol, 150 μl de C6F6) : c'est la série no 2.
L'éthanol de maïs a été utilisé comme référence.
2.2 Acquisitions RMN–2H
Le spectre RMN–2H quantitatif des échantillons a été mesuré dans des conditions standard, selon le protocole décrit dans les publications antérieures [5,20] et dans les méthodes officielles [21] : α = 90° soit P1 = 10 μs, TR = 6,9 s, SW = 1200 Hz.
Seule la séquence d'impulsion a été modifiée, afin de faire intervenir le signal électronique ERETIC dans le spectre.
Le spectromètre utilisé est un Bruker DRX 500 de fréquence nominale 500,13 MHz en proton, équipé d'une sonde adaptée à la mesure isotopique en 2H (sonde 10 mm 2H–1H, lock 19F). Le spectromètre comporte trois canaux radiofréquence, permettant à la fois l'observation des noyaux 2H, le découplage 1H et l'injection du signal ERETIC.
Après une multiplication exponentielle (LB = 2 Hz) et une transformation de Fourier sur 64 K, les surfaces des raies ont été déterminées par ajustement du spectre à un modèle lorentzien à l'aide du logiciel PERCH (PERCH NMR Software © Université de Kuopio, Finlande).
Pour chaque tube, quatre spectres ont été réalisés.
2.3 ERETIC
La technique ERETIC consiste à générer électroniquement un signal radiofréquence, émis par une antenne annexe et recueilli lors de l'acquisition du FID de l'échantillon. Dans notre cas, lors des acquisitions de RMN–2H, le signal ERETIC est émis par la bobine proton par l'intermédiaire d'un coupleur RF (Fig. 1) ; lors des acquisitions de RMN–1H, ce signal est émis par la bobine 2H [14,15].
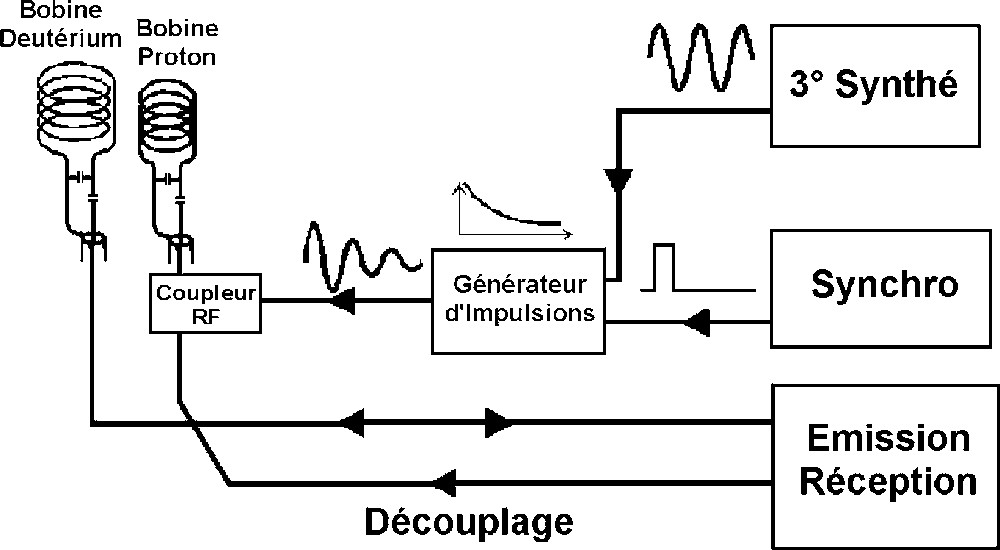
Dispositif de création d'ERETIC.
Après transformation de Fourier, ce signal donne naissance à un pic supplémentaire sur le spectre (Fig. 2).

Spectre 2H découplé 1H de l'éthanol, pour un tube contenant de la TMU, en présence du signal électronique ERETIC.
2.4 Mesure des temps de relaxation longitudinale T1
Pour les deux séries de tubes, les temps de relaxation longitudinale T1 ont été mesurés, sur chaque site du spectre, par la séquence d'inversion–récupération (12 temps d'inversion compris entre 5 ms et 8 s). Le traitement des données est effectué par un programme dédié au calcul des temps de relaxation, intégré à la console du spectromètre.
2.5 Mesure du rapport signal/bruit
Pour chaque spectre, le rapport signal/bruit a été mesuré sur le plus petit pic quantifié du spectre: CH3–CHD–OH, le bruit étant mesuré entre 2,4 et 2,7 ppm.
2.6 Calcul des Ai
La mesure des concentrations respectives des différents composés a été effectuée à partir des spectres 1H quantitatifs, selon un protocole publié précédemment [22] : α = 10°, TR = 9 s, SW = 10 ppm.
2.6.1 Méthode de la référence interne
Sur le spectre RMN–2H obtenu sur un échantillon contenant le composé étudié et une référence interne, les surfaces :
| (4) |
Sur le spectre RMN–1H obtenu sur le même échantillon, les surfaces :
D'où :
| (5) |
2.6.2 Méthode ERETIC
Les spectres RMN–2H et RMN–1H sont obtenus à partir des échantillons à mesurer et sur un tube de référence contenant le composé étudié dont les abondances isotopiques Airef sont connues. (Dans le cas de ce travail, le tube de référence contenait un alcool de maïs dont les abondances isotopiques étaient :A1ref=106,4±0,7ppm et A2ref=119,8±1,0ppm).
Le spectre 2H obtenu sur le tube de référence fournit les aires :
Un spectre 2H obtenu sur le tube à mesurer fournit :
Le spectre 1H obtenu sur le tube de référence fournit :
Le spectre 1H obtenu sur le tube à mesurer fournit :
| (6) |
| (7) |
À partir des quatre spectres 1H acquis sur chaque tube, une valeur moyenne pour est obtenue. Cette valeur est utilisée pour calculer les Ai pour chaque spectre 2H. À partir des quatre spectres 2H acquis sur chaque tube, une valeur moyenne et un écart type sont calculés.
3 Résultats et discussion
Les valeurs de T1 obtenues pour les deux séries de tubes sont données dans le Tableau 1. Dans la première série de tubes (protocole classique), le T1 de la TMU est du même ordre de grandeur que ceux des sites de l'éthanol et le T1 le plus grand est égal à 1,3 s (CHD). L'absence de TMU dans la deuxième préparation ne modifie que peu les T1 des sites de l'éthanol et le T1 le plus grand est égal à 1,2 s (CH2D).
Temps de relaxation longitudinale (T1 en s) pour les différents sites des spectres 2H de l'éthanol et du TMU pour les deux séries de tubes
| Site du spectre 2H | CH3–CHD–OH | CH2D–CH2–OH | TMU |
| Série no 1 (avec TMU) | 1,3 | 1,1 | 1,2 |
| Série no 2 (sans TMU) | 1,1 | 1,2 | — |
Le T1 le plus long (T1max) détermine le temps de répétition TR nécessaire entre chaque scan pour obtenir un spectre quantitatif (en effet, pour une précision ≥ 0,7%, il faut TR ≥ 5 × T1max). Le T1max étant très similaire entre les deux séries de tubes, le TR est identique pour les deux séries d'acquisitions. En revanche, avec la méthode ERETIC, le T1 de la référence n'est plus à prendre en compte dans le calcul du TR pour une acquisition quantitative. Pour d'autres types d'échantillons, par exemple les lipides, le T1 de la TMU est largement supérieur à celui des sites mesurés. La présence de TMU allonge alors considérablement le TR. Pour ce type de composés, la méthode ERETIC (dans les conditions du deuxième protocole, en absence de TMU) diminuerait notablement le TR et donc la durée totale d'expérience.
Tous échantillons confondus, la moyenne du rapport signal/bruit est de 180 pour la première préparation (avec TMU) et de 290 pour la deuxième préparation (éthanol quasiment pur). Le rapport signal/bruit a été multiplié, entre la première et la seconde série de tubes, par un facteur d'environ 1,6. La sensibilité est souvent un paramètre limitant en RMN–2H. Or, grâce à la méthode ERETIC, l'absence de référence interne dans le tube induit une augmentation de la concentration. En conséquence, à nombre de scans (donc à temps d'acquisition) constant, le rapport signal/bruit est amélioré et l'écart type de la mesure est réduit. À rapport signal/bruit constant, une réduction du temps d'expérience est donc possible. Pour les échantillons étudiés, le temps d'expérience aurait été divisé par environ deux. Ce gain pourrait être largement supérieur, pour des échantillons dont la co-solubilité échantillon–référence nécessite une dilution dans un solvant.
Les abondances isotopiques spécifiques mesurées par les deux méthodes (ERETIC et référence interne) sur les deux séries de tubes (avec et sans TMU) sont données dans le Tableau 2 et représentées graphiquement sur les Figs. 3 et 4. En routine, la précision de mesure des Ai, nécessaire à l'authentification des éthanols, est de 1%. Pour les deux séries de tubes, les valeurs de Ai sont toutes non significativement différentes à l'écart type de la mesure près (≤ 1%). Avec les deux protocoles, la référence ERETIC permet donc de caractériser les échantillons avec la précision standard requise.
Valeurs des Ai (en ppm) des différents éthanols, avec les deux types de référence et, dans le cas de la référence ERETIC, pour les deux séries de tubes (valeurs moyennes et écarts types obtenus à partir de quatre mesures)
| Valeur par TMU (série no 1) | écart type | Valeur par ERETIC (série no 1) | écart type | Valeur par ERETIC (série no 2) | écart type | ||
| Vin | A1 | 100,7 | 0,4 | 101,0 | 0,6 | 100,8 | 0,4 |
| A2 | 125,7 | 0,7 | 125,7 | 1,2 | 123,7 | 1,2 | |
| Betterave | A1 | 90,4 | 0,6 | 89,6 | 0,7 | 89,8 | 0,4 |
| A2 | 119,0 | 0,6 | 117,7 | 1,0 | 116,8 | 1,0 | |
| Canne | A1 | 109,4 | 0,3 | 108,8 | 0,5 | 110,5 | 0,3 |
| A2 | 123,4 | 0,6 | 122,4 | 1,2 | 123,3 | 0,4 | |
| Blé | A1 | 109,7 | 0,5 | 108,8 | 1,1 | 109,9 | 0,3 |
| A2 | 127,8 | 1,5 | 126,4 | 1,9 | 127,6 | 0,3 |
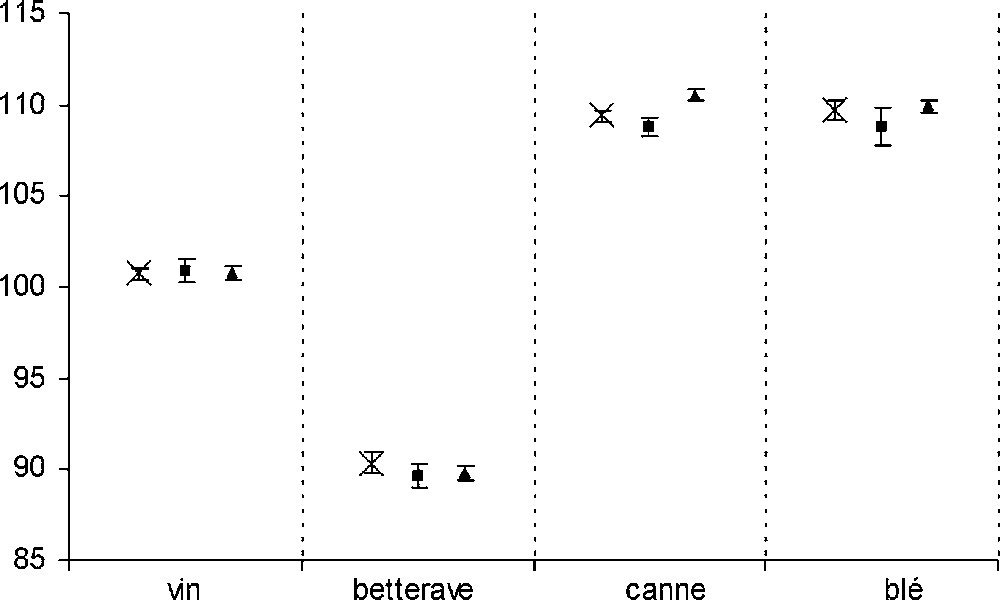
Valeurs de A1 (en ppm) des éthanols avec les deux types de références et, dans le cas de la référence ERETIC, pour les deux séries de tubes : (×) référence TMU (série no 1) ; (■) référence ERETIC (série no 1) ; (▴) référence ERETIC (série n °2). Les barres d'erreur sont placées à ± l'écart type.
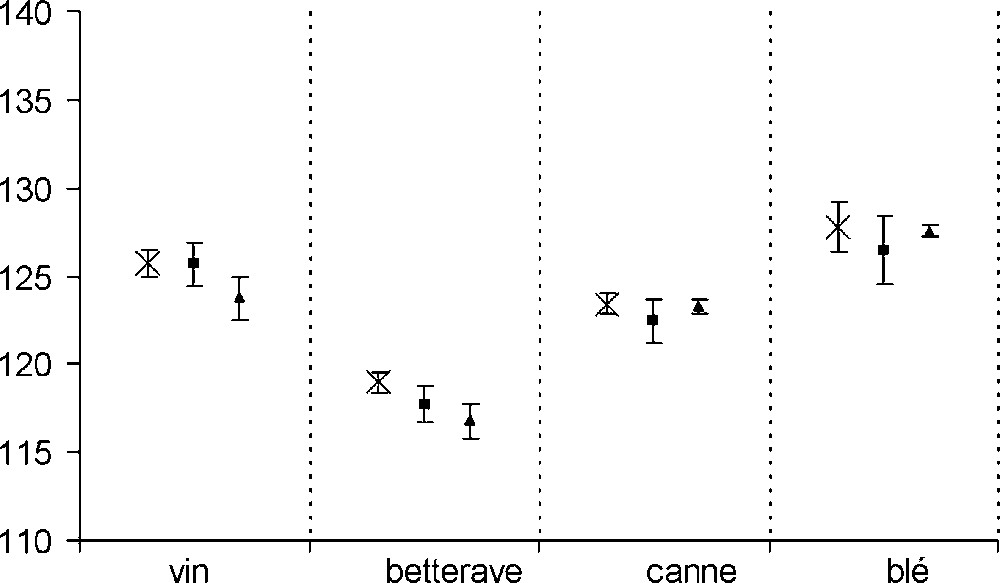
Valeurs de A2 (en ppm) des éthanols avec les deux types de référence et, dans le cas de la référence ERETIC, pour les deux séries de tubes : (×) référence TMU (série no 1) ; (■) référence ERETIC (série no 1) ; (▴) référence ERETIC (série no 2). Les barres d'erreur sont placées à ± l'écart type.
Pour les deux séries d'expériences, l'écart type de la mesure est globalement similaire pour ce qui concerne la référence interne et ERETIC, et inférieur à 1%. L'utilisation de la référence ERETIC ne semble donc pas diminuer la précision de la mesure. L'examen du Tableau 2 tend à montrer que la précision est un peu meilleure sur la seconde série de tubes. Toutefois, dans cette expérience, les écarts types étant calculés sur un petit nombre de points (quatre spectres), il est donc difficile d'interpréter finement l'évolution de ce paramètre.
Dans les deux méthodes de référence, les concentrations de produits contenus dans les tubes doivent être parfaitement connues. Les concentrations peuvent s'exprimer :
La référence interne ne fait intervenir qu'un seul tube dans la mesure (Vt = V′t) et l'évaluation des concentrations relatives peut donc se réduire à des mesures de masses et de puretés :
| (8) |
En revanche, la méthode ERETIC utilise une référence placée dans un tube différent de celui de l'échantillon mesuré. Si la méthode gravimétrique ci-dessus est employée, les comparaisons inter-tubes font intervenir dans le calcul les volumes totaux des différents tubes :
| (9) |
Les volumes totaux étant techniquement très difficiles à maîtriser avec la précision exigée, le choix a donc été fait de mettre en œuvre les spectres 1H pour la mesure des concentrations relatives (équation (6)). La durée de ces spectres 1H est très courte (1 min) et n'allonge que très peu la durée totale de l'expérience : les tubes 10 mm, avec leur forte concentration, fournissent une quantité de signal largement suffisante dès 16 scans (signal/bruit ≈ 3000).
Pour obtenir la meilleure précision, une calibration d'ERETIC est souhaitable sur un tube référence, à chaque session d'expérience. Cependant, dans le cas d'une mesure sur une série d'échantillons, ceci n'allonge que peu le temps d'expérience.
Le choix d'une référence interne pour la RMN quantitative 2H est toujours difficile. Non seulement le composé choisi doit satisfaire de nombreux critères (solubilité, volatilité, hygroscopicité, stabilité dans le temps, inertie chimique, non-recouvrement de raies avec l'échantillon et temps de relaxation de l'ordre de ceux de l'échantillon), mais idéalement, il doit être identique pour tous les composés qui doivent être comparés (dans une étude de filiation isotopique par exemple). De plus, son abondance isotopique doit être parfaitement connue. Peu de molécules satisfont à tous ces critères. À notre connaissance, les seuls composés pour lesquels l'abondance isotopique deutérium a été certifiée par une procédure de validation internationale sont : l'eau (échelle V.SMOW) [2] et la TMU [23]. L'eau est peu soluble dans les solvants organiques et la TMU a un T1 qui est très souvent nettement plus long que ceux de l'échantillon, dès que celui-ci comporte des molécules de poids moléculaire supérieur à 100 g mol–1. Tout ces éléments ne sont plus à prendre en compte dans le cas de la méthode ERETIC. De plus, la référence électronique ne pollue pas l'échantillon et permet donc une récupération plus facile de l'échantillon pour d'autres analyses.
Dans les applications de la méthode ERETIC à la détermination de concentrations [15–18], une « concentration équivalente » est affectée au signal de référence par calibration sur un tube qui contient un composé de concentration connue. Ici, le signal ERETIC est utilisé comme un standard de travail. Il n'est pas nécessaire de lui affecter une concentration et/ou une abondance isotopique équivalente. Les propriétés de ce signal sont uniquement utilisées pour corriger les variations de sensibilité qui ne manquent pas d'intervenir entre la mesure de la référence et celle de l'échantillon. La référence a, dans le cas de notre étude, les mêmes caractéristiques physico-chimiques que l'échantillon (nature, concentration). Les temps de relaxation de la référence et de l'échantillon peuvent donc être considérés comme identiques. Il n'est donc plus indispensable de se placer dans des conditions quantitatives strictes. Ce point n'a pas été abordé dans ce travail et devra être vérifié expérimentalement, mais il pourrait conduire à une réduction supplémentaire du temps d'expérience après optimisation du temps de répétition et de l'angle de nutation.
4 Conclusion
Cette étude démontre que la référence électronique ERETIC permet d'obtenir en SNIF–NMR des résultats comparables à ceux obtenus avec la méthode de référence interne. En effet, l'écart entre les deux résultats est inférieur à la précision (1%) nécessaire à l'authentification des échantillons.
Bien que nécessitant une calibration préalable et l'intervention d'un spectre 1H, le protocole mis en place avec l'utilisation d'ERETIC est avantageux sur plusieurs points : pas de problème de choix de la référence (co-solubilité, pas de superposition de pics), pas de contamination ou d'interaction avec l'échantillon, élimination des coûts induits par l'utilisation d'une référence chimique calibrée, simplification de la préparation des tubes (pas de pesées), augmentation du rapport signal/bruit à nombre de scans constant et réduction éventuelle du temps de répétition.
Cette méthode est actuellement appliquée au laboratoire à l'étude isotopique d'échantillons dont le spectre est plus complexe que celui de l'éthanol.