

1 Introduction
Les produits alimentaires sont des matrices reconnues pour leur complexité de structure et de composition. De plus, nombre d'entre eux présentent une hétérogénéité spatiale à de multiples échelles, du niveau moléculaire au niveau macroscopique. Parallèlement, ils sont le siège de transferts interne et externe qui leur confèrent une instabilité qu'il est nécessaire de maîtriser, ou tout au moins de contrôler, pour garantir leur qualité. Aussi, les techniques qui peuvent permettre d'accéder à des informations sur la structure et la composition dans des conditions dynamiques représentent-elles un enjeu important pour la recherche agroalimentaire ; l'imagerie de résonance magnétique (IRM) est l'une de ces techniques. L'IRM est, depuis de nombreuses années, unanimement reconnue dans le domaine médical comme un outil puissant pour le diagnostic et la réalisation d'études fonctionnelles. Malgré la richesse des informations accessibles par IRM, le développement de cette technique pour l'étude des biomatériaux en général et des produits alimentaires en particulier est encore limité. En 1992, lors du deuxième Congrès international de la RMN appliquée en agroalimentaire, des études IRM avaient été présentées et laissaient prévoir un rapide développement de cette technique en science des aliments. Paradoxalement, à ce jour, le constat est modeste, et le développement escompté n'a pas été aussi rapide.
L'explication réside en grande partie dans les spécificités scientifiques requises pour la réalisation de mesures IRM sur des produits alimentaires par rapport au domaine médical. En effet, le diagnostic médical repose sur une analyse qualitative basée sur le contraste des images IRM. Par opposition, en agroalimentaire, une analyse quantitative des images est souvent incontournable. Pour conduire cette analyse quantitative, différentes stratégies sont possibles.
L'analyse peut être réalisée directement à partir des niveaux de gris des images lorsque des informations morphologiques sont nécessaires. Par exemple, pour la quantification de défauts dans des tissus végétaux ou la répartition des tissus adipeux au sein de carcasse de porc. L'analyse quantitative peut être aussi conduite sur des images paramétriques, T2, T1, ou des coefficients de diffusion. Enfin celle-ci peut être faite par une transformation des images IRM en cartographies de la teneur en eau, de la porosité ou de la quantité de glace.
Au-delà de l'extraction du critère quantitatif, il est aussi nécessaire de valider l'interprétation des images IRM. En effet, suivant les mécanismes participant à l'établissement du contraste des images, ou plus généralement à l'origine des variations spatiales du signal RMN, les voies d'action pour modifier les caractéristiques des produits seront différentes. Le contraste peut être potentiellement expliqué, soit par des variations macroscopiques de densité protonique, ou des variations de teneur en eau ou en matière grasse, soit par des modifications microscopiques de la structure des matrices, ou enfin par des modifications à l'échelle moléculaire. Aussi, pour chaque examen IRM de produits alimentaires, il est nécessaire d'associer une étude complète des signaux de relaxation par des mesures RMN des paramètres de relaxation, afin de valider l'interprétation des images et pour faciliter la définition des protocoles de mesure IRM. L'objectif de cet article est de montrer comment cette démarche a permis de faire de l'IRM une technique puissante d'investigation pour la recherche agroalimentaire. Pour un approfondissement des applications agroalimentaires de la RMN et de l'IRM, nous recommandons la lecture de l'ouvrage de B. Hills [1].
La Fig. 1 présente des exemples d'images IRM de produits alimentaires. Un simple examen visuel de ces images permet, dans certains cas, de proposer une interprétation pertinente de l'image IRM. Par exemple, dans le cas de la tomate, on reconnaît très bien la structure interne de la tomate, avec les loges, les pépins, le placenta, le péricarpe (Fig. 1a). De la même façon, l'image IRM de la carcasse de poulet est cohérente avec l'anatomie déjà connue par ailleurs (Fig. 1b). En revanche, quelle interprétation proposer pour expliquer la zone blanche proche de la surface dans l'image de la pomme ? S'agit-il d'un gradient de teneur en eau, d'une modification locale de la structure, ou d'une inhomogénéité liée à l'antenne de réception de l'équipement (Fig. 1c) ? Cette situation se retrouve de façon plus aiguë lors de l'examen de produits manufacturés et complexes, tels que des fromages, pour lesquels une analyse anatomique n'est pas courante (Fig. 1d).
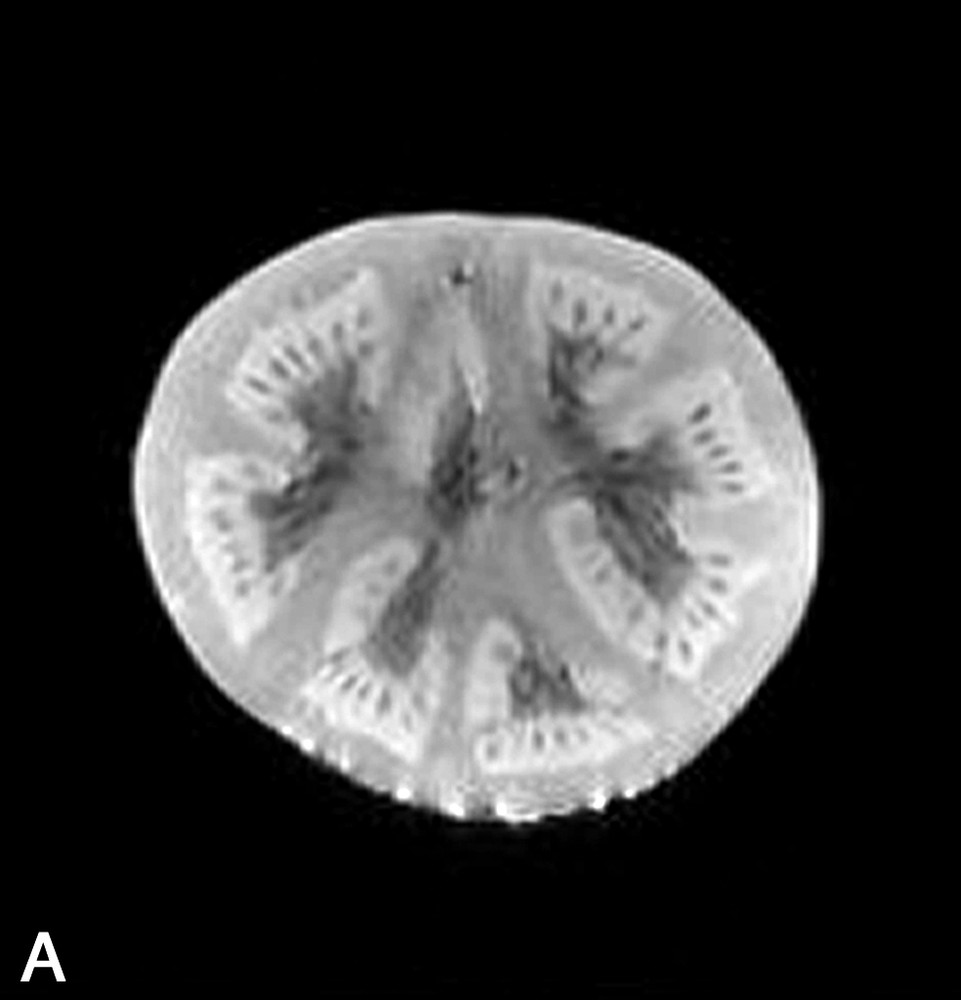
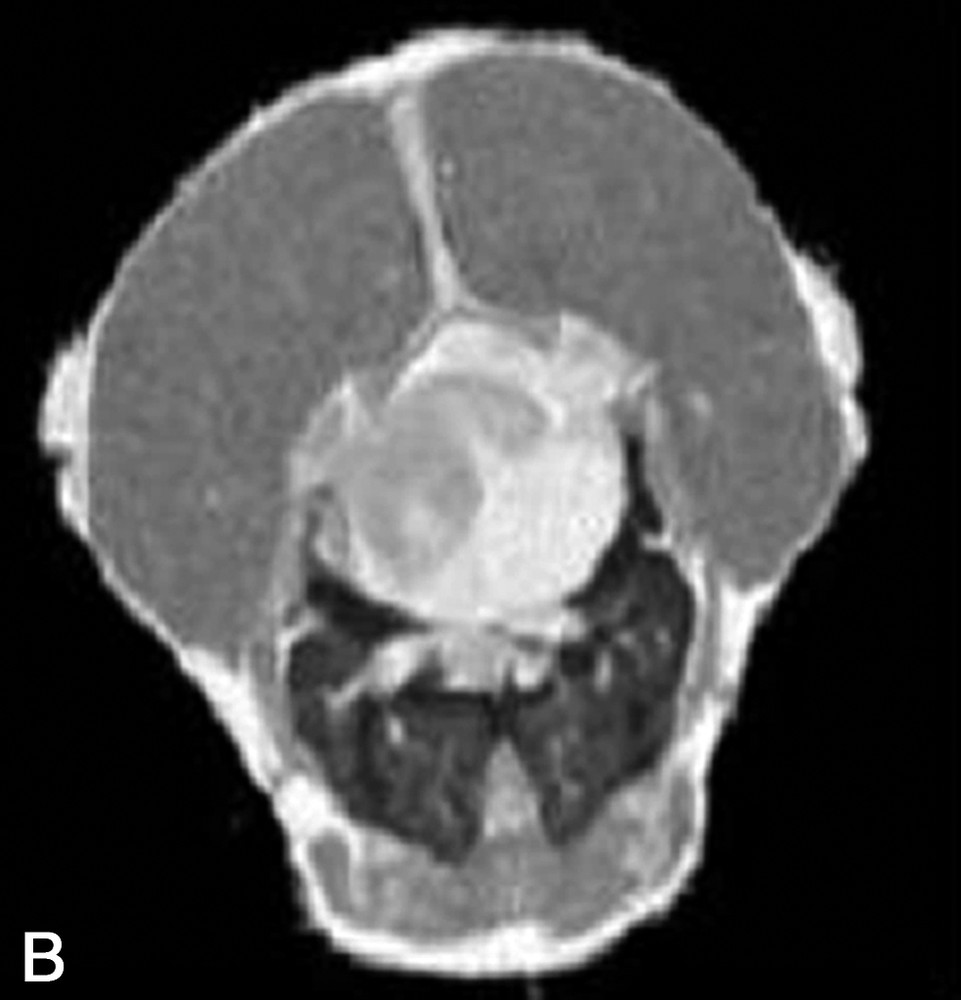
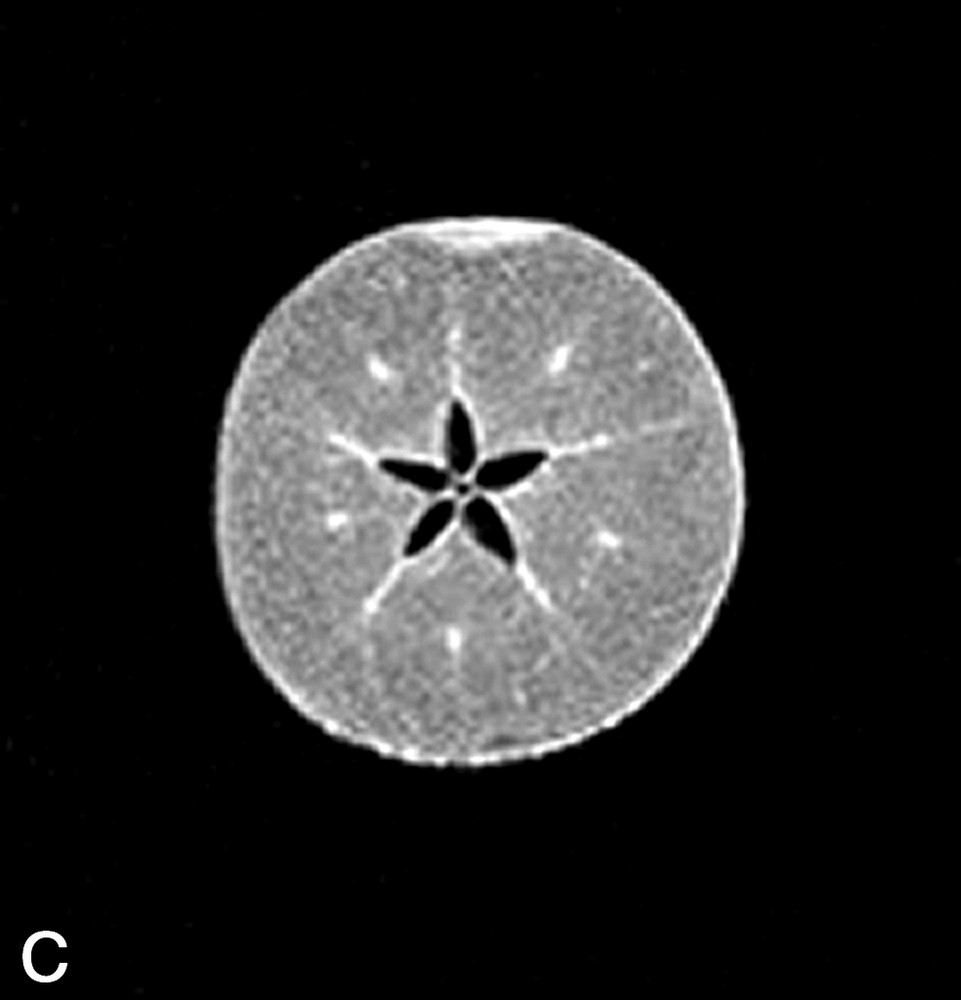
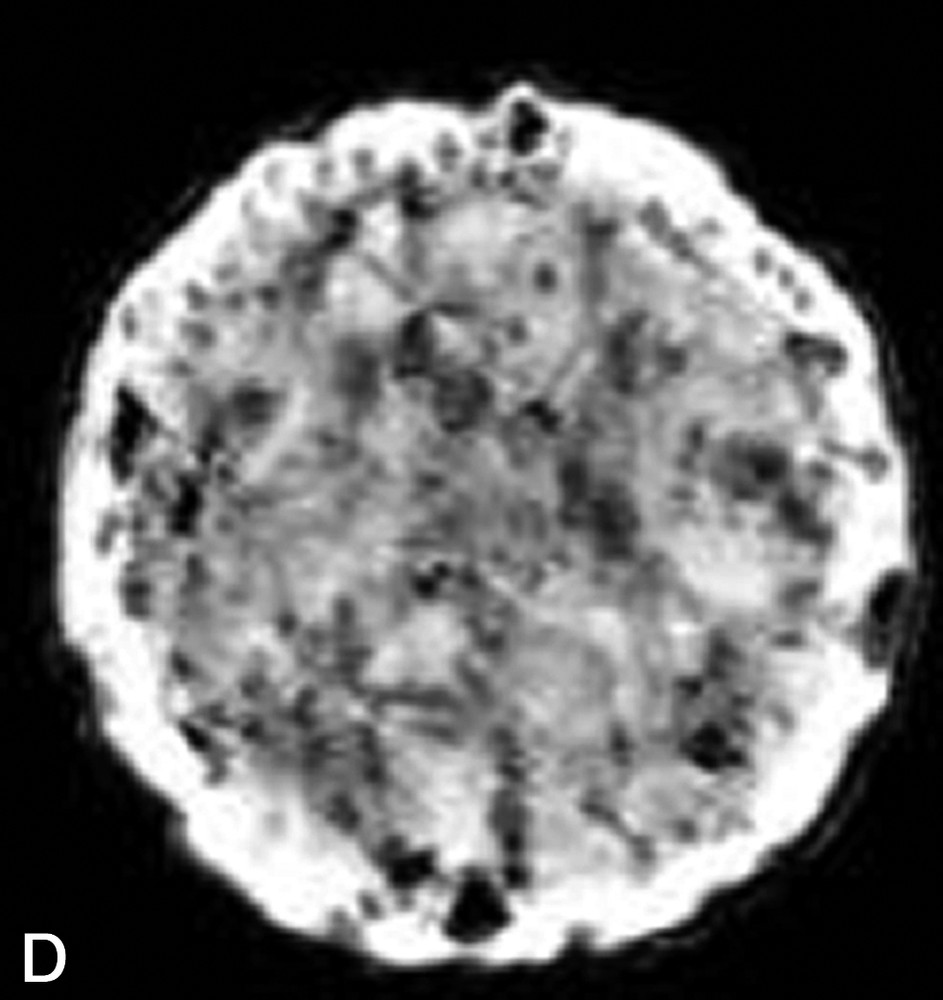
(a) Image IRM d'une tomate acquise avec une séquence d'écho de spin à 0,2 T (Magnetom Open Siemens). L'épaisseur de coupe était de 3 mm et la résolution de 0,7 mm. (b) Image IRM d'un poulet acquise avec une séquence d'écho de spin à 0,2 T (Magnetom Open Siemens). L'épaisseur de coupe était de 10 mm et la résolution de 1 mm. (c) Image IRM d'une pomme acquise avec une séquence d'écho de spin à 0,2 T (Magnetom Open Siemens). L'épaisseur de coupe était de 3 mm et la résolution de 0,7 mm. (d) Image IRM d'un fromage acquise avec une séquence d'écho de gradient à 0,2 T (Magnetom Open Siemens). L'épaisseur de coupe était de 4 mm et la résolution de 0,7 mm.
En effet, l'origine du contraste dépend, d'une part, de paramètres expérimentaux tels que la séquence d'impulsion ou les paramètres associés (temps d'écho, temps de récupération, angle de bascule...) et, d'autre part, de paramètres intrinsèques des tissus, tels que la densité protonique et les temps de relaxation. À leur tour, ces paramètres intrinsèques RMN dépendront de la composition chimique, de la structure microscopique et de la structure moléculaire et seront donc pour l'expérimentateur autant de moyens pour mesurer les caractéristiques physico-chimiques des produits.
2 La densité protonique
La densité protonique ρ est le paramètre RMN qui conduit à des images dont l'intensité du voxel est la plus directement interprétable. En effet, la densité protonique est seulement sensible à la composition des échantillons, par opposition aux paramètres de relaxation, qui dépendront à la fois de la composition et de la structure. Plus précisément, la densité protonique renseigne sur le volume de molécule protonée contenu dans un voxel. L'application la plus directe des images en densité de proton concerne la détection de cavités remplies d'air, ou de tout autre gaz, au sein des produits. Ces cavités peuvent être des bulles dans des mousses, des trous dans des fromages ou des fruits, ou des fractures dans des produits transformés. Suivant le rapport entre la taille de la cavité et la résolution de l'image IRM, la démarche de mesure sera différente. Ainsi, trois situations peuvent être rencontrées. (1) Si la taille de la cavité est supérieure à la résolution IRM, une simple acquisition, avec une séquence d'écho de spin, permet la détection de la cavité. Par exemple, des images en densité protonique ont été exploitées pour mesurer la taille et la répartition des trous dans des fromages [2–5]. Dans ce cas, une segmentation de l'image permet d'extraire, d'une part, le nombre de trous et, d'autre part, de calculer des caractéristiques de tailles et de formes. Ces caractéristiques sont utilisées comme paramètres pour distinguer différentes technologies fromagères ou permettre un classement des fromages suivant des indices de qualité [5]. En effet, des trous de forme régulière seront attribuables à la libération du CO2 résultant de la fermentation microbienne, tandis que des trous de formes irrégulières correspondront à des défauts ou accidents de fabrication. D'autres exemples concernent l'observation des défauts internes des fruits provoqués par le développement de parasite, ou la formation de crevasses ou de fractures au sein des produits à la suite de l'application de traitements technologiques tels que le séchage ou la cuisson [6–9]. (2) Si la taille de la cavité est de l'ordre de grandeur de la résolution, une séquence d'écho de gradient pourra être appliquée. En effet, l'écart de susceptibilité magnétique entre l'eau et l'air induit des déformations locales des lignes de champ magnétique et le signal IRM est artificiellement atténué (Fig. 2). Cet effet n'est pas corrigé par une acquisition avec une séquence d'écho de gradient. De plus, l'effet sera d'autant plus élevé que l'intensité de B0 sera grande. Cette démarche permet ainsi de diminuer le seuil de détection de l'IRM pour les petites cavités, mais interdit une mesure précise des caractéristiques de taille [4]. Par la suite, les images peuvent être analysées, soit par une méthode de segmentation, soit par des méthodes d'analyse de texture [10]. (3) Si la taille de la cavité est inférieure à la résolution de l'image IRM, il n'est pas possible de mesurer des caractéristiques morphologiques. Néanmoins, la présence de micro-bulles induit une atténuation du signal IRM par effet de volume partiel. L'intensité du signal devient donc proportionnelle au rapport volumique total des bulles dans le voxel. Cet effet a été exploité pour l'étude de la stabilité des mousses au drainage [11]. En revanche, cette application nécessite une correction a posteriori de l'intensité de l'image par la relaxation spin–spin de la phase dispersante, qui varie en fonction du drainage. En effet, la relation entre l'intensité du signal IRM S(x) et la densité protonique est donnée par la relation suivante :
| (1) |
| (2) |
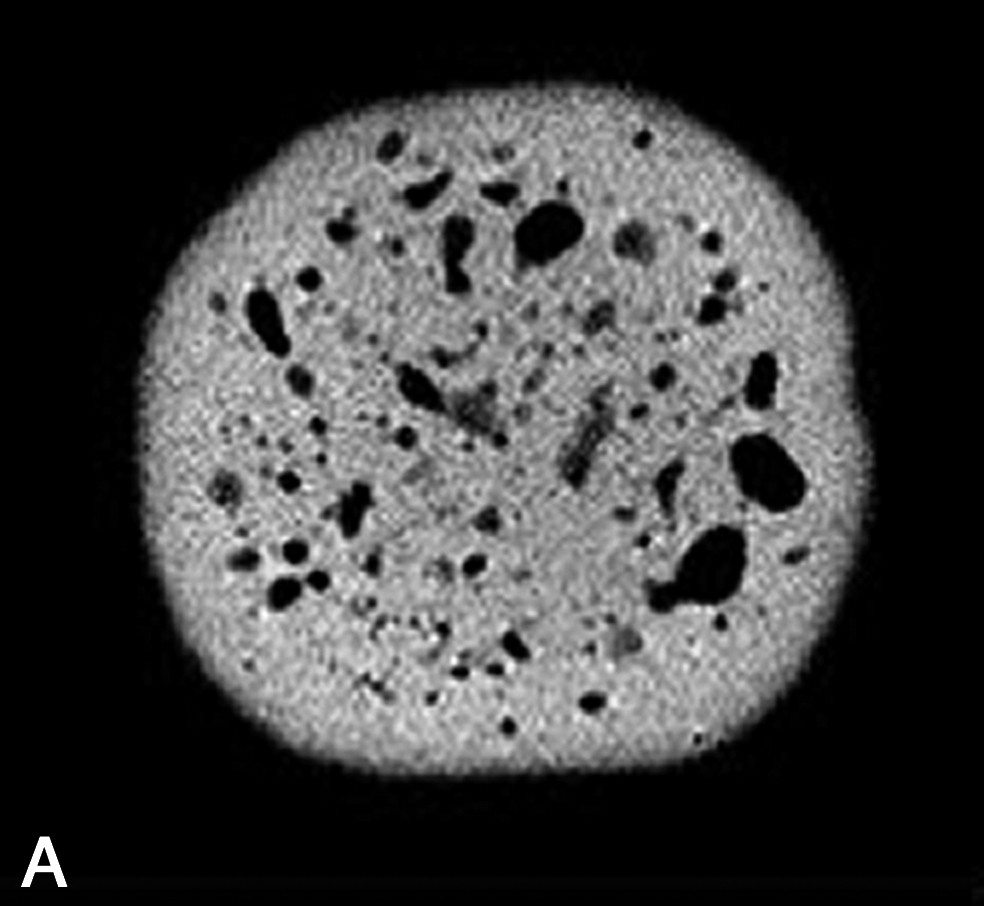
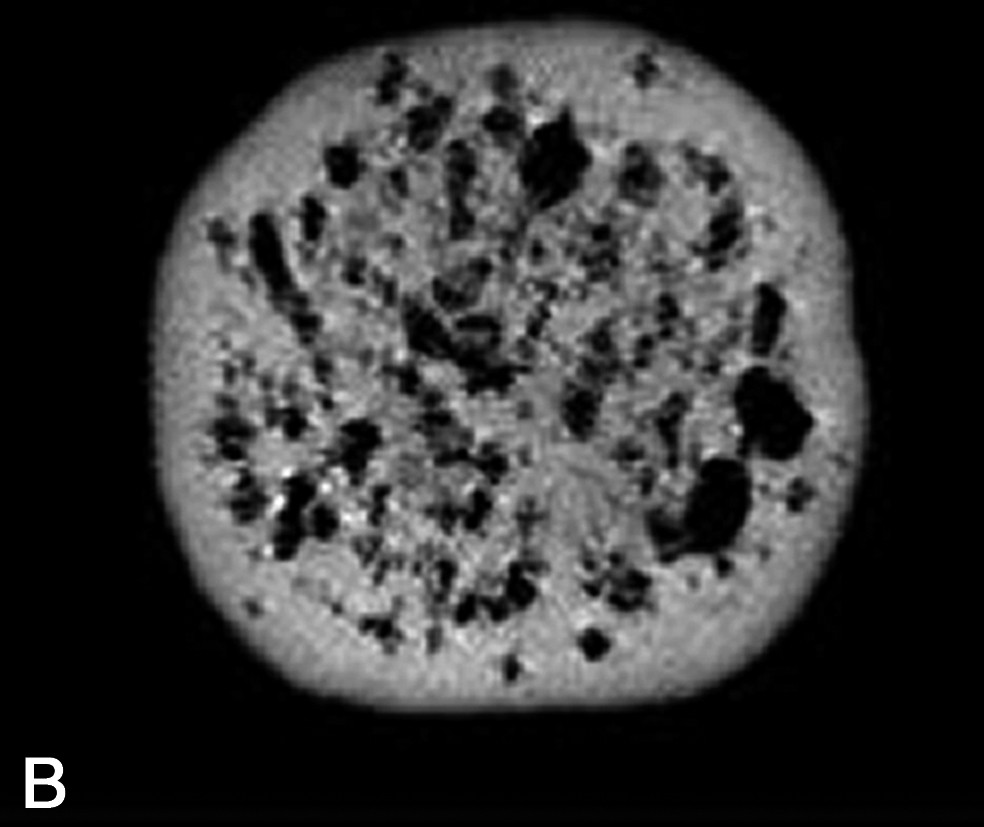
Image IRM de fromage acquise à 0,2 T (Magnetom Open Siemens). Comparaison entre une séquence d'écho de spin (à gauche ; TE = 15 ms, TR = 1500 ms) et une séquence d'écho de gradient (à droite ; TE = 23 ms et TR = 1500 ms). L'épaisseur de coupe était de 4 mm et la résolution de 0,7 mm.
Pour minimiser les effets de la relaxation spin–spin, soit un temps d'écho suffisamment court est fixé, soit le temps de relaxation est mesuré et introduit dans l'équation (2).
Outre leur utilisation pour la détection de cavités, les images de densité de proton ont été utilisées pour la mesure de la teneur en eau ou en matière grasse. Une correction de l'intensité des images IRM par le temps de relaxation spin–spin est souvent nécessaire, car celui-ci varie fortement avec la teneur en eau. L'utilisation de cette démarche est particulièrement adaptée pour la mesure de la teneur en eau au cours de l'application de procédé de séchage ou de réhydratation [12–21]. Si les produits ne contiennent pas de matière grasse, une relation linéaire entre le signal IRM corrigé et la teneur en eau est ainsi généralement trouvée. En revanche, si les produits contiennent de la matière grasse, même à faible concentration (2% par exemple), le modèle n'est plus linéaire pour les faibles valeurs de la teneur en eau. Le temps de relaxation de l'eau diminue fortement et le signal de la matière grasse liquide devient important [13]. Le signal IRM S(x) dépendra alors de la teneur en matière grasse et non de la teneur en eau. Aussi est-il nécessaire de vérifier au préalable les valeurs des temps de relaxation de l'eau pour définir les conditions d'acquisition IRM, en particulier le temps d'écho, afin d'éviter des erreurs dans l'estimation de la teneur en eau. D'une façon générale, cette démarche n'est possible que pour des produits maigres ou avec très peu de matière grasse ; dans le cas contraire, les méthodes basées sur la mesure des déplacements chimiques seront plus adaptées [22,23].
3 Les temps de relaxation
Deux paramètres de relaxation sont classiquement exploités comme vecteurs d'information en IRM : le temps de relaxation spin–spin (T2) et le temps de relaxation spin–réseau (T1). La relaxation T2 présente, par rapport à la relaxation T1, une sensibilité supérieure à la composition et à la structure des produits alimentaires. Aussi ce paramètre est-il retenu dans la majorité des cas. La relaxation T1 sera plutôt utilisée comme un traceur de changement de température au sein des produits.
3.1 Relaxation et composition chimique
Dans les produits fortement hydratés, les temps de relaxation sont d'abord affectés par la composition des produits, et plus particulièrement par la teneur en eau. L'exploitation du temps de relaxation T2 comme traceur de la teneur en eau suppose la réalisation au préalable d'un modèle de calibration sur l'ensemble de la gamme de variation de teneur en eau. Ce modèle de calibration permettra d'obtenir des cartographies de teneur en eau à partir des cartographies de T2. Cette démarche permet d'obtenir des valeurs de teneur en eau plus précises, car la sensibilité par rapport à une mesure de la densité protonique est supérieure. De plus, le calcul des temps de relaxation conduit à une correction des inhomogénéité des images IRM, qui n'est pas toujours prise en compte lors de la mesure de la densité protonique. Ainsi, des profils de teneur en eau dans des pâtes au cours la réhydratation [24] et des cartographies 2D de la teneur en eau au sein de gel laitier en cours d'égouttage [25,26] ont été mesurés. L'égouttage est une étape importante intervenant dans la fabrication du fromage. Elle permet d'amener le lait, préalablement coagulé, à la teneur en eau requise pour un affinage correct. La déshydratation du gel de lait dit « caillé » résulte de la contraction naturelle du gel, qui favorise alors une expulsion du sérum. La Fig. 3 est un exemple de cartographie de teneur en eau directement issue d'une cartographie de T2. Le caillé se trouve au sein d'un récipient cylindrique à la base duquel est placé un filtre qui permet l'écoulement de la phase aqueuse expulsée. Un récipient est placé en dessous pour récupérer la phase aqueuse. Au cours de l'égouttage, un gradient de teneur en eau s'établit entre le centre et la périphérie du caillé. La contraction naturelle du caillé accélère l'égouttage et conduit à une expulsion de la phase aqueuse. Une partie de celle-ci est récupérée dans le récipient, tandis qu'une autre partie se trouve localisée à la surface du caillé. Au cours du temps, elle s'écoulera au travers du gel et accentuera le gradient de teneur en eau du caillé. Pour ce type de matrice, un simple modèle linéaire relie la teneur en eau et la vitesse de relaxation (Fig. 4). Ainsi, la mesure se déroule en trois étapes, acquisition IRM par une séquence multi-écho, puis calcul des temps de relaxation et enfin conversion du temps de relaxation en teneur en eau. Soulignons que le modèle linéaire n'est adapté que pour la détermination de la teneur en eau au sein du caillé et non pour celle de la teneur en eau de la phase aqueuse exsudée. D'autres travaux se sont intéressés à l'étude de l'effet de la congélation/décongélation sur la répartition de l'eau au sein de caillé (Fig. 5) [27].
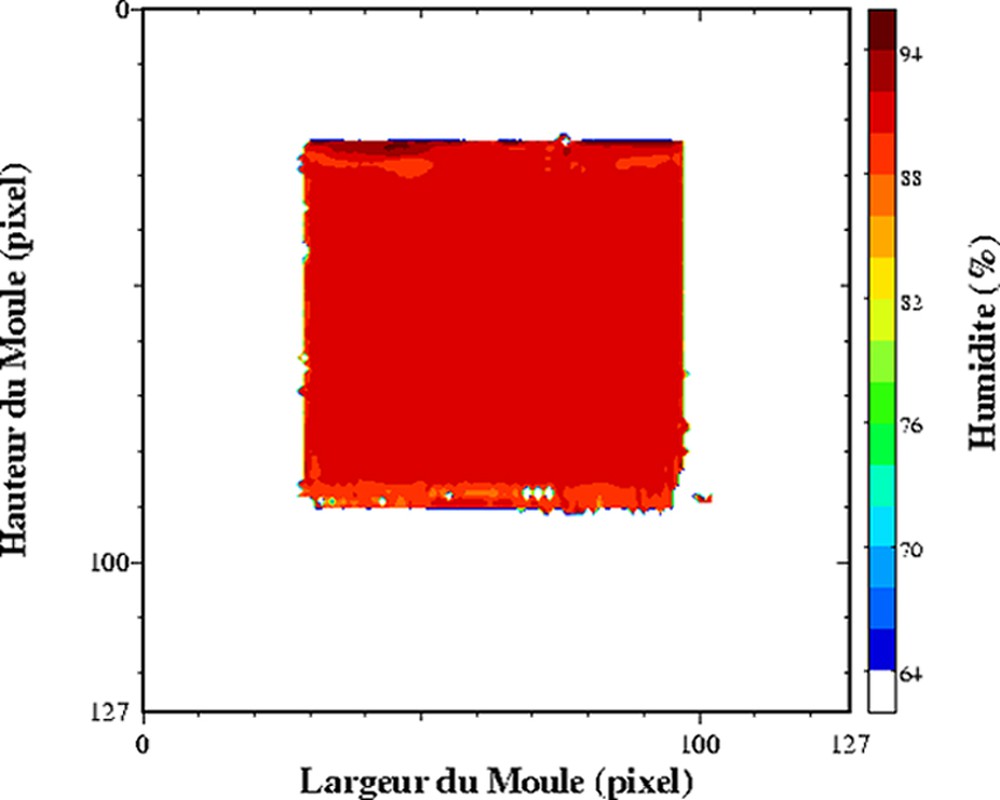
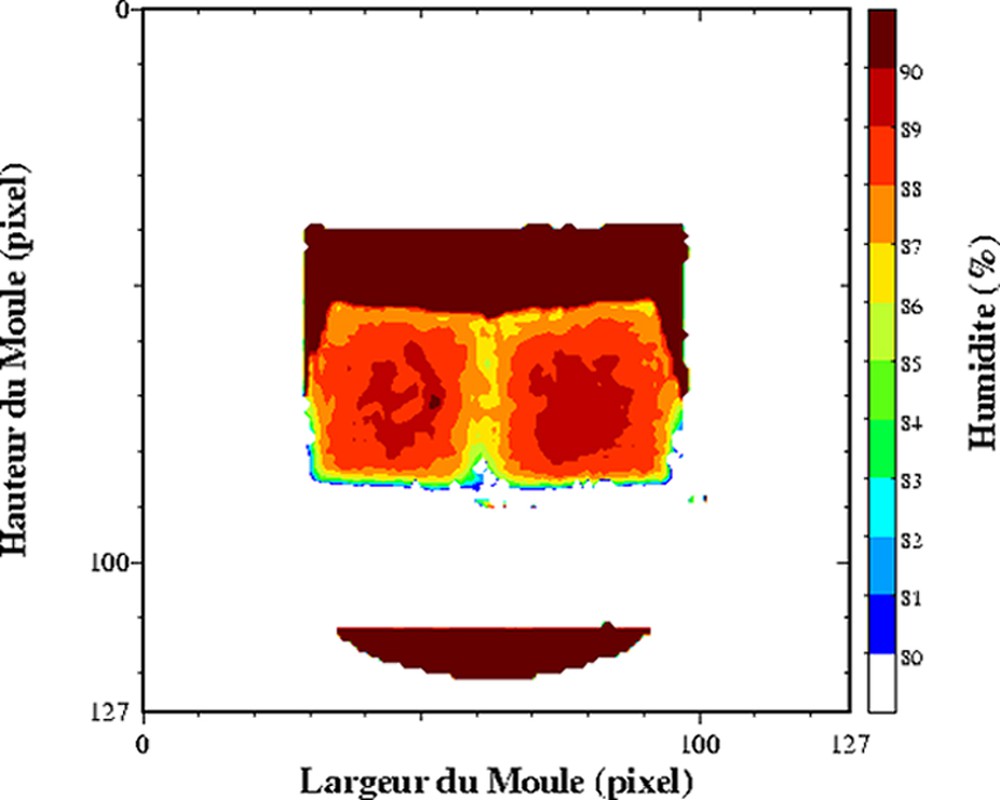
Cartographie de l'humidité d'un lait coagulé en cours d'égouttage. Acquisition à T = 0 (à gauche) et à T = 2 h 20 après le début de l'égouttage (à droite). Séquence multi-écho n × TE = 17, nombre d'échos, n = 24, TR = 1500 ms. L'épaisseur de coupe était de 4 mm et la résolution de 1,4 mm [25].
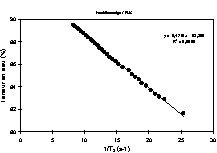
Corrélation entre la vitesse de relaxation 1/T2 et la teneur en eau d'un gel laitier [25].
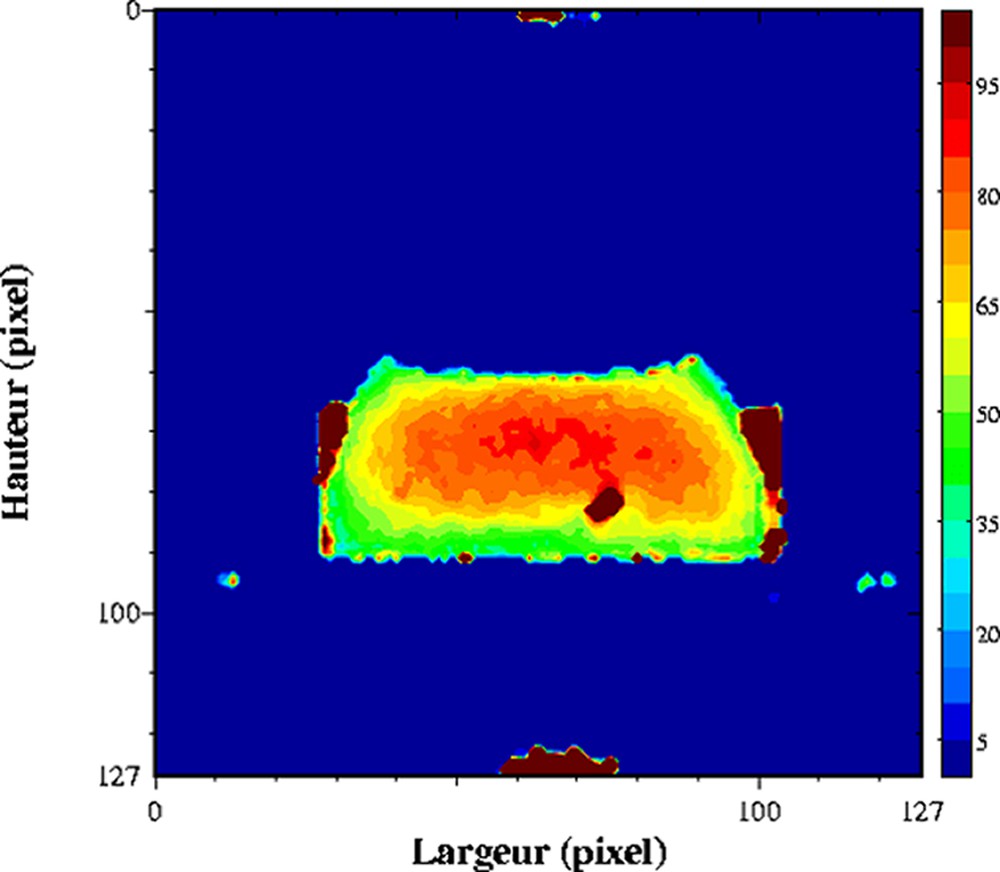
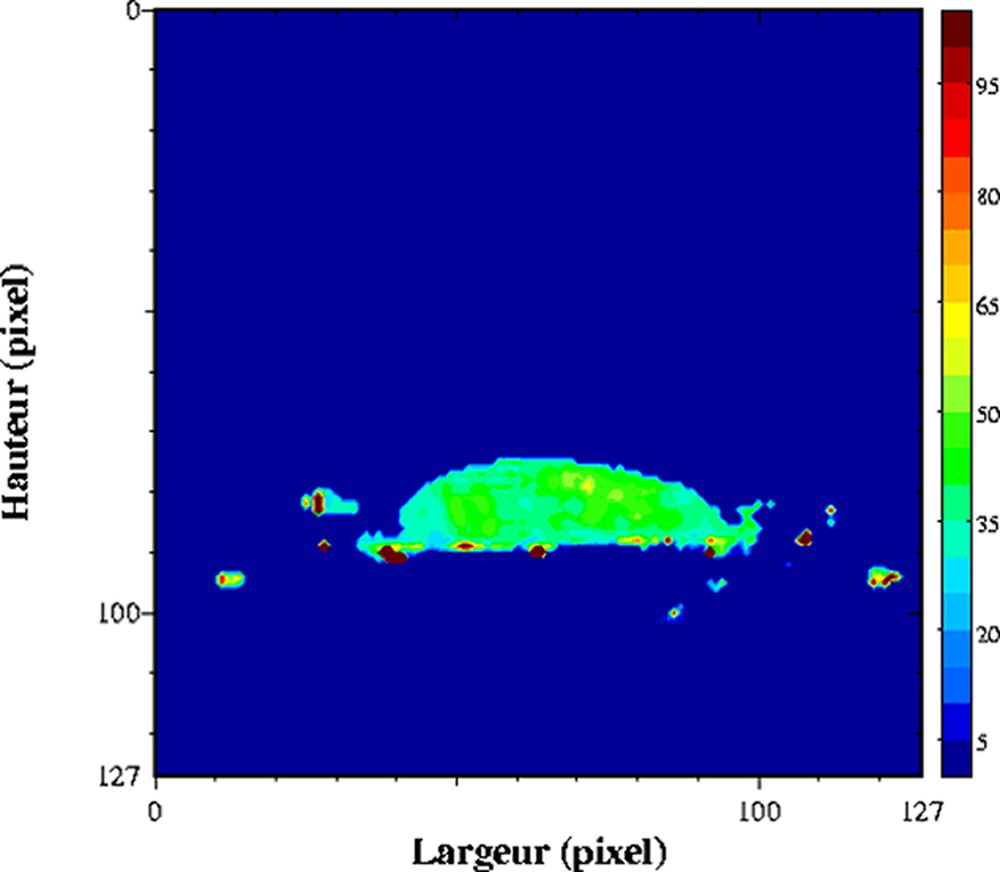
Influence de la congélation/décongélation sur un gel laitier. Cartographie de T2 acquise à 0,2 T (Magnetom Open, Siemens) avec une séquence multi-écho, n × TE = 17, nombre d'échos, n = 24, TR = 1500 ms. L'épaisseur de coupe était de 4 mm et la résolution de 1,4 mm [26].
3.2 Relaxation de l'eau et structure microscopique
La relaxation de l'eau dans les produits alimentaires suit généralement un comportement mono-exponentiel. Le temps de relaxation calculé décrit un comportement moyen de l'ensemble des molécules d'eau. Ce temps de relaxation a souvent été interprété par le modèle à deux sites de Zimmerman et Brittin [28]. Ce modèle considère que les molécules d'eau se répartissent entre l'eau dite « libre » et l'eau dite « liée », chaque population pouvant être décrite par un temps de relaxation spécifique. Du fait de l'existence d'échanges diffusionnels rapides entre les deux populations (P), le temps de relaxation observé est donné par :
| (3) |
Ce modèle est actuellement fortement critiqué, car il ne considère qu'un seul mécanisme pour expliquer la diminution de la relaxation de l'eau en fonction de la concentration des autres molécules [29]. Néanmoins, l'effet des échanges diffusionnels sur la relaxation reste vérifié et conditionne le comportement mono ou multi-exponentiel de la relaxation. Si les échanges diffusionnels entre différents compartiments deviennent lents, alors le signal de relaxation sera décrit par une multi-exponentielle. Dans certains cas, l'attribution des temps de relaxation permet d'estimer les quantités d'eau dans chacun des compartiments. La limite de l'échange diffusionnel sera dépendante d'une part de la distance entre les sites hydriques différents (a) et de l'écart de vitesse de relaxation de l'eau dans les deux sites (ΔR2) [30]:
| (4) |
D étant le coefficient de diffusion de l'eau. Ainsi, différentes situations peuvent favoriser un échange diffusionnel lent : par exemple, des gels possédant des micro-hétérogénéités de teneur en eau, soit en raison d'un défaut de mélange des ingrédients, soit en raison d'un traitement physico-chimique particulier. Par exemple, les traitements thermiques et mécaniques des gels de protéines laitières peuvent favoriser la formation d'une micro exsudation ou synérèse [31]. La synérèse correspond à l'expulsion de la phase aqueuse à la suite de la contraction naturelle des gels laitiers. Celle-ci se traduit par l'apparition de micro-fissures remplies d'eau. Le signal de relaxation ne devient pas nécessairement bi-exponentiel, car la quantité d'eau dans ces micro-fissures n'est pas importante. Toutefois, une analyse continue du temps de relaxation met en évidence une augmentation de la largeur de la distribution de la relaxation de l'eau (Fig. 6). Cette augmentation de la distribution décrit donc une hétérogénéité de structure au sein du gel provoquée par l'apparition de ces micro-fissures. Ce comportement se traduit sur l'image IRM par une forte variabilité des temps de relaxation (Fig. 7). Du fait de la différence de temps de relaxation, une image pondérée en T2 permet de mettre en évidence l'hétérogénéité du gel. Si le phénomène s'accentue, la relaxation de l'eau sera bi-exponentielle et il devient possible de suivre au cours du temps le re-largage de la phase aqueuse hors du gel [32,33]. La composante de relaxation dont le temps de relaxation est le plus court sera attribuable à la relaxation de l'eau confinée dans le gel poreux, tandis que celle dont le temps de relaxation sera proche de celui de la phase aqueuse correspondra à la relaxation de l'eau re-larguée. Les populations respectives des deux exponentielles seront proportionnelles à la quantité d'eau dans le gel, et à la quantité d'eau re-larguée. La différence de temps de relaxation est telle entre les deux composantes qu'il est alors possible par IRM de ne visualiser que la fraction de l'eau non exsudée ou exsudée, et de faciliter ainsi la quantification et la localisation des fractions au cours du temps (Fig. 8). Cet exemple illustre l'intérêt de l'IRM pour la caractérisation des propriétés des gels alimentaires en matière de rétention de l'eau.

Effet du traitement thermique sur la distribution des T2 d'un gel laitier obtenu par ultrafiltration. La distribution T2 du produit sans traitement (•) est élargie par le traitement thermique (△). Les mesures RMN ont été obtenues par une séquence CPMG avec un spectromètre à 0,47 Tesla (Minispec PC120, Bruker). Les mesures RMN ont été triplées [65].

Image IRM pondérée T2 d'un gel laitier acquise par une séquence d'écho de spin (TE = 26 ms, TR = 1500 ms) à 0,2 T (Magnetom Open, Siemens). L'épaisseur de coupe était de 4 mm et la résolution de 0,7 mm. Les voxels en hyper-signal correspondent à des zones de fracture remplies de phase aqueuse [65].

Image IRM pondérée T1 d'un gel laitier en cours d'égouttage et préalablement découpé en cube. L'image IRM a été acquise par une séquence d'écho de spin (TE = 15 ms, TR = 500 ms) à 0,2 T (Magnetom Open, Siemens). L'épaisseur de coupe était de 4 mm et la résolution de 0,7 mm. Les voxels en hypo-signal correspondent aux espaces entre les cubes de gel dans lesquels la phase aqueuse est expulsée.
Le comportement multi-exponentiel de la relaxation de l'eau est aussi observé pour les produits végétaux. En effet, l'organisation interne des cellules conduit à un comportement tri-exponentiel de la relaxation. Chacune des composantes de relaxation peut être attribuée à différentes fractions d'eau : l'eau dans la paroi cellulaire, dans le cytoplasme et dans la vacuole (Tableau 1) [34–36]. Les échanges diffusionnels entre ces compartiments hydriques étant lents par rapport à la relaxation, la condition pour un comportement multi-exponentiel est alors respectée. En revanche, si la vitesse d'échange est modifiée par une altération des parois des tissus, alors la relaxation en sera fortement affectée. La sensibilité de la relaxation à la structure microscopique des tissus cellulaires a été exploitée dans de nombreux travaux, tels que l'étude de la maturation et de la croissance [37,38], l'étude des effets des traitements thermiques [39,40] ou l'étude de défauts internes [41–48]. L'IRM peut être utilisée aussi pour l'étude de la transformation des produits végétaux [49,50]. Par exemple, l'analyse conjointe RMN et IRM des signaux de tranches de pomme de terre ayant subi un traitement de friture a permis de démontrer l'effet du traitement sur la structure cellulaire [51]. Le Tableau 2 décrit les temps de relaxation d'une pomme de terre crue. Les trois composantes de relaxation ont été attribuées, d'une part, à la relaxation de l'eau en interaction avec les grains d'amidon (T2 = 4 ms), à la relaxation de l'eau du cytoplasme (T2 = 184 ms) et à la relaxation de l'eau dans la vacuole (T2 = 563 ms). La cartographie IRM T2 d'une pomme de terre permet de ne calculer qu'un temps de relaxation moyen (Fig. 9). Les T2 varient entre 540 ms au centre et 360 ms en périphérie. Cette hétérogénéité est à rapprocher de la structure de la pomme de terre, dont le centre est constitué de cellules riches en eau et pauvres en amidon : la moelle. L'application d'un traitement de friture d'une minute induit de fortes variations des temps de relaxation (Fig. 10). En particulier, le temps de relaxation au centre a fortement augmenté. Cet effet résulte de l'augmentation de la température et des contraintes mécaniques subies par les tissus, qui conduisent à un éclatement des cellules et au re-largage du liquide intracellulaire. La condition d'échange diffusionnel rapide est modifiée et le temps de relaxation de l'eau augmente. Le même mécanisme intervient au sein de la pomme lorsque l'intégrité des cellules est endommagée par une action mécanique en surface. L'influence de la diffusion sur le signal a été démontrée en comparant l'intensité du signal acquis par un écho de Hahn et par une séquence multi-écho (CPMG) [41].
Temps de relaxation (ms) et amplitude (%) calculés à partir de la courbe de relaxation spin–spin d'une pomme [35]. Les écart types sont calculés avec cinq répétitions
| Composante de relaxation | T2i (ms) | Ai (%) |
| Eau dans la paroi | 57 ± 5,3 | 3,7 ± 0,5 |
| Eau dans le cytoplasme | 404 ± 4 | 12,5 ± 1,8 |
| Eau dans la vacuole | 1184 ± 46 | 83,8 ± 1,3 |
Temps de relaxation (ms) et amplitude (%) calculés à partir de la courbe de relaxation spin–spin d'une pomme de terre crue [50]. Les écarts types sont calculés avec cinq répétitions
| Composante de relaxation | T2i (ms) | Ai (%) |
| Eau dans le grain d'amidon | 4 ± 1 | 12 ± 4 |
| Eau dans le cytoplasme | 184 ± 32 | 27 ± 13 |
| Eau dans la vacuole | 563 ± 51 | 61 ± 17 |
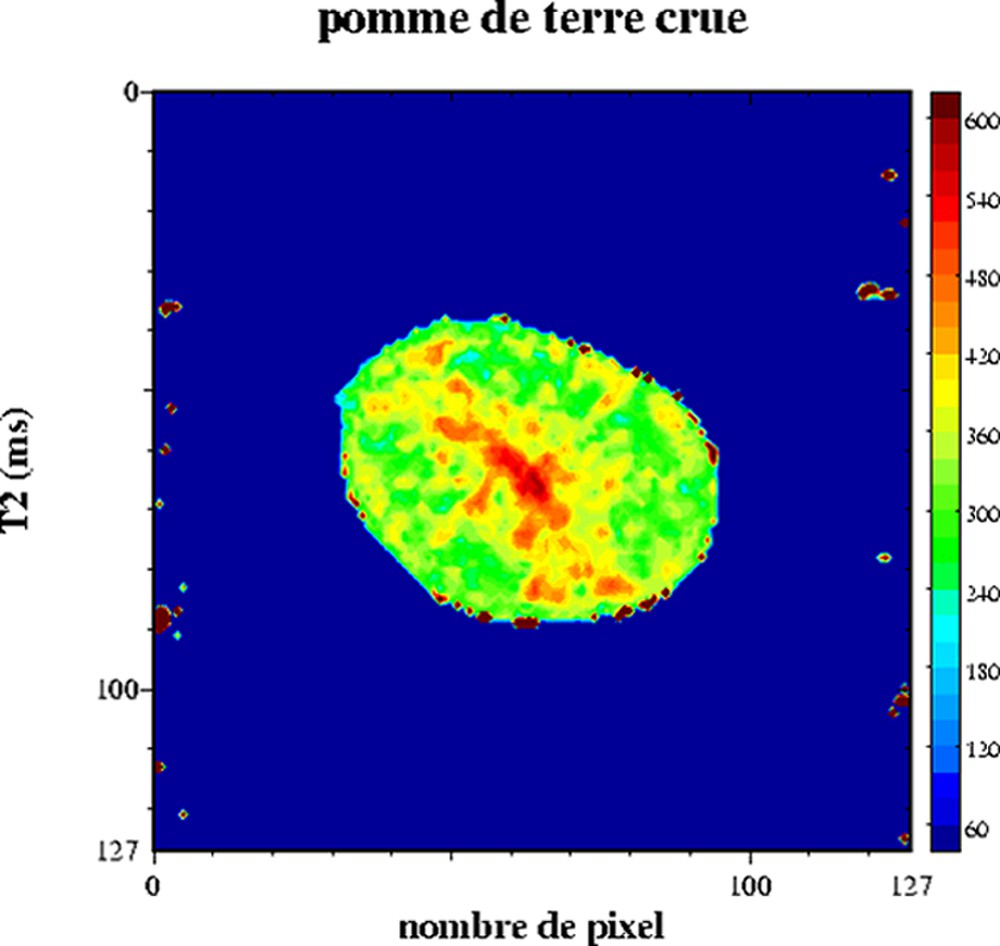
Cartographie T2 d'une pomme de terre crue. Séquence multi-écho n × TE = 17, nombre d'échos, n = 24, TR = 1500 ms, acquise à 0,2 T (Magnetom Open, Siemens). L'épaisseur de coupe était de 3 mm et la résolution de 1,2 mm [50].
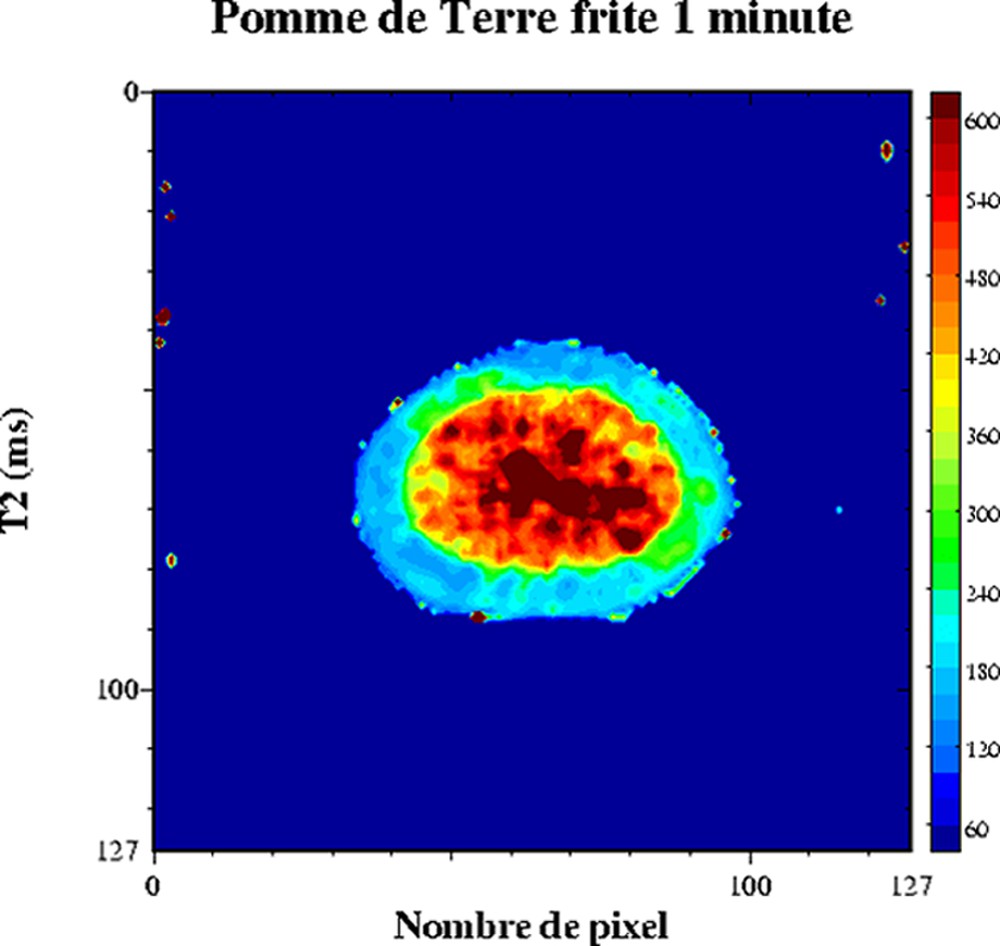
Cartographie T2 d'une pomme de terre frite (1 min). Séquence multi-écho n × TE = 17, nombre d'échos, n = 24, TR = 1500 ms). L'épaisseur de coupe était de 3 mm et la résolution de 1,2 mm [50].
3.3 Relaxation et structure moléculaire
Les temps de relaxation RMN sont des paramètres dynamiques qui renseignent sur la mobilité des structures moléculaires en présence. La gamme de variation du temps de relaxation spin–spin est en effet très importante lorsque la molécule d'intérêt passe de l'état liquide à l'état cristallisé, par exemple. Grâce à cette sensibilité, la relaxation RMN est devenue la méthode de référence pour la quantification de la quantité de cristaux de matière grasse dans des margarines [52]. Depuis, la RMN est couramment utilisée par les industriels du secteur des margarines. Plus récemment, cette méthode a été transposée à l'IRM pour l'étude de la cristallisation de la matière grasse. Dans ce cas, le signal IRM obtenu par une simple séquence d'écho de spin correspond au signal de la fraction liquide de la matière grasse. La relaxation de la fraction cristallisée étant trop rapide pour que le signal soit détectable par IRM (T2 = 15 μs, Fig. 11), les variations de niveau de gris sont directement corrélées à la quantité de matière grasse liquide. L'avantage de la technique réside dans la possibilité d'accéder aux cinétiques de cristallisation de produits de grand volume et donc de se rapprocher des conditions industrielles [53–56] et, d'autre part, d'obtenir des cartographies de taux de cristallisation dans des produits transformés [57] ou directement dans des pièces de porcs [58]. Ce dernier travail a démontré que les tissus adipeux étaient structurés en couches caractérisées par des points de fusions différents. Des prélèvements dans chacune de ces couches ont démontré l'existence d'une composition triglycéridique différente, et un triglycéride spécifique qui expliquait à lui seul plus de 96% des variations des quantités de matière grasse cristallisée a été identifié. Les perspectives actuelles de ces travaux concernent l'étude des facteurs d'alimentation en interaction avec les facteurs de race ou de sexe des animaux.
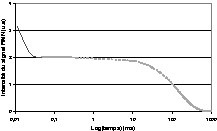
Signal de relaxation RMN incluant la relaxation de la fraction cristallisée (—) et la relaxation de la fraction liquide (----).
Cette même démarche peut être appliquée à l'étude des phénomènes de congélation [59,60]. En effet, le temps de relaxation de la glace est trop faible pour pouvoir être mesuré, et l'intensité du signal mesurée après un écho est directement proportionnelle à la fraction non congelée de l'eau. Ainsi, les cinétiques et les fronts de congélation ont été mesurés au cours de la décongélation des caillés de fromagerie [27], ou au cours de la congélation de cylindre de pommes de terre [61].
3.4 Relaxation de l'eau et structure moléculaire des macromolécules
Le temps de relaxation de l'eau dans les produits fortement hydratés peut en effet être décrit par le modèle dit à trois sites de Fedotov et al. [62].
Ce modèle considère trois catégories de protons : (a) les protons de l'eau « libre », dont la mobilité ne dépend pas de celle des macromolécules du milieu ; (b) les protons de l'eau d'« hydratation », dont la mobilité est affectée par la présence d'autres molécules ; (c) les protons qui s'échangent entre les molécules du milieu et les molécules d'eau (protons échangeables des fonctions OH, NH2, et SH des protéines ou des sucres). Cet échange est appelé « échange chimique », par opposition à l'échange diffusionnel, qui décrit des échanges de molécules d'eau entre des environnements différents. La relaxation observée est alors décrite par [63,64] :
| (5) |
| (6) |
De nombreux auteurs ont montré que le terme était faible devant le terme et donc que les échanges chimiques étaient le mécanisme dominant de la relaxation de l'eau [29,63–65]. La relaxation de l'eau devient donc, via les mécanismes d'échange chimique, une sonde des changements de structure des molécules impliquées dans ces échanges. Par conséquent, tout facteur chimique ou physique qui conduira à une modification de la mobilité des molécules pourra être potentiellement appréhendé par la relaxation de l'eau. Par exemple, le temps de relaxation a été exploité pour l'étude de l'affinage des fromages [66]. En effet, au cours de l'affinage, le pH augmente, ce qui provoque une modification importante de la structure des protéines et, par voie de conséquence, de la texture du produit. À pH acide, le fromage est friable et dur, tandis qu'il devient souple et plus mou à pH basique. Une étude RMN a montré que le temps de relaxation de l'eau était particulièrement sensible aux changements de structure des protéines provoqués par la remontée du pH (Fig. 12) ; le temps de relaxation devenait alors un excellent traceur de suivi de l'affinage. L'intérêt de l'utilisation de l'IRM était, d'une part, de permettre de suivre le même fromage au cours de l'affinage et, d'autre part, de construire ainsi des itinéraires d'évolution des fromages en fonction de la composition initiale ou en fonction des variables du procédé (température de hâloir, hygrométrie etc.). La Fig. 13 représente des images IRM de fromages à différents stades de l'affinage. Ces images sont des cartographies des temps de relaxation T2. Les voxels de faibles valeurs de T2 correspondent à la zone non-affinée avec un pH bas (≈ 5). Les voxels de T2 = 55–60 ms sont attribuables à la zone affinée avec un pH de 7 à 7,5. Un simple seuillage de la cartographie des T2 permet de mesurer les surfaces relatives de ces deux zones et d'en déduire des cinétiques d'évolution en fonction de la composition initiale des fromages. L'utilisation du temps de relaxation de l'eau comme traceur des changements de conformation des macromolécules peut être généralisée à de nombreuses opérations technologiques, telles que la coagulation thermique des blancs d'œuf [67], la friture de pomme de terre [36], ou la congélation/décongélation des produits carnés [68–71].
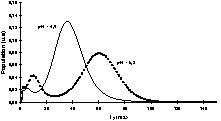
Effet du pH sur la distribution des T2 de l'eau d'un fromage en cours d'affinage. Les mesures RMN ont été obtenues avec une séquence CPMG avec un spectromètre à 0,45 T (Minispec PC120, Bruker) [65].
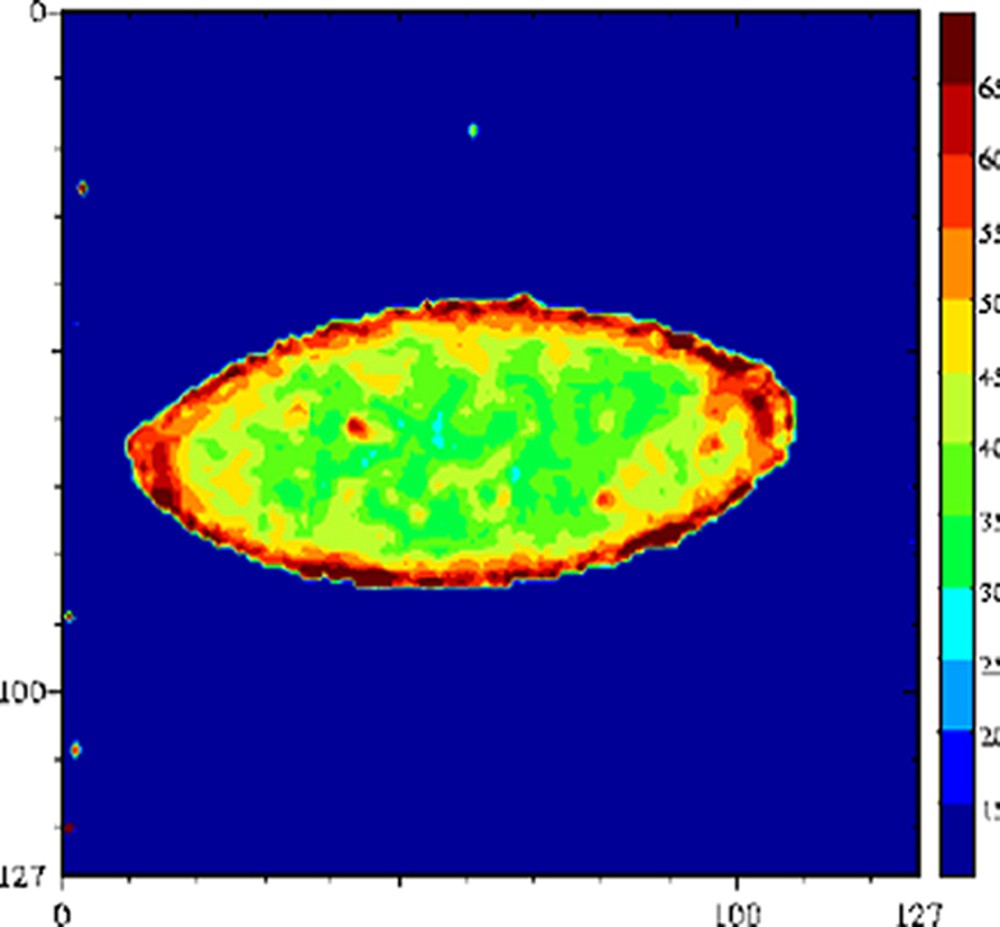
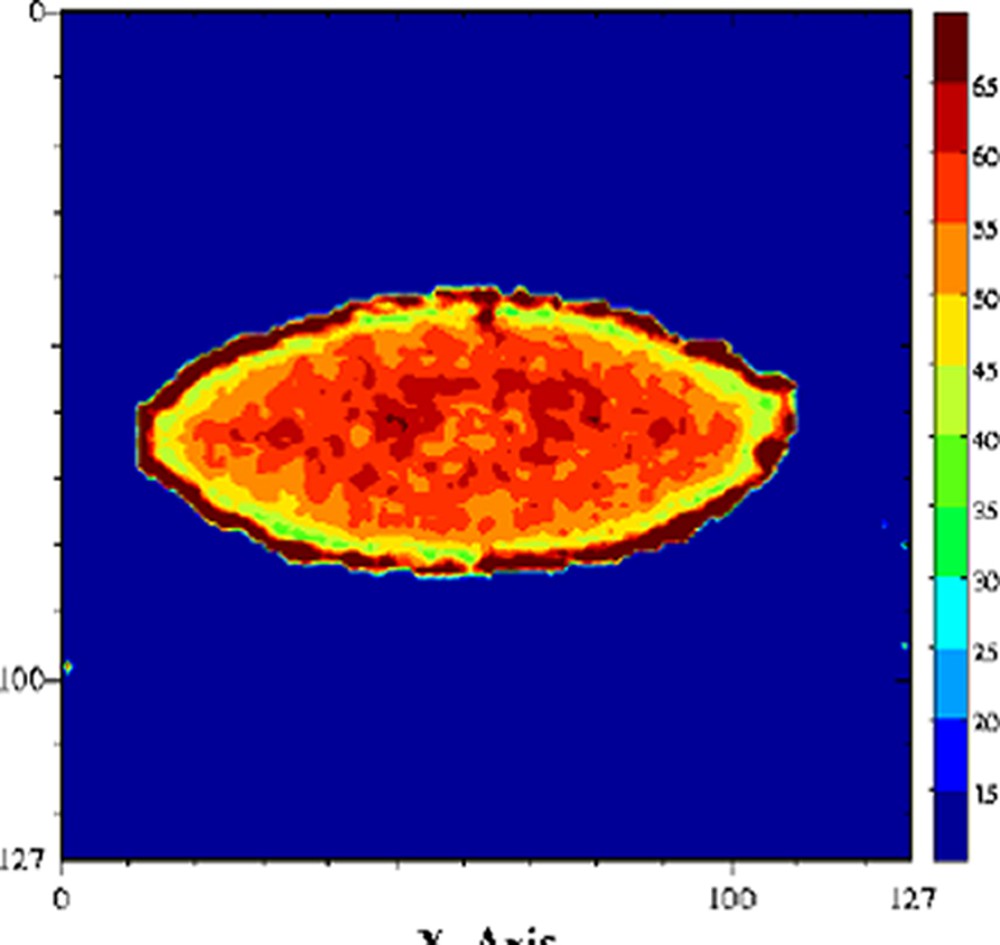
Cartographie T2 d'un fromage à pâte molle au cours de l'affinage, stade jeune (gauche) et complètement affiné (droite). Séquence multi-écho n × TE = 17, nombre d'échos, n = 24, TR = 1500 ms, acquise à 0,2 T (Magnetom Open, Siemens). L'épaisseur de coupe était de 4 mm et la résolution de 1,4 mm [65].
4 Conclusion
Les potentialités de la technique IRM pour la recherche en science des aliments et en génie des procédés sont remarquables. Parmi les techniques spectrométriques disponibles pour l'étude des matrices alimentaires, l'IRM est une des rares à associer à la fois des caractéristiques techniques autorisant des mesures non destructrices et en dynamique et des caractéristiques métrologiques permettant d'accéder à des informations sur la composition et sur la structure. Grâce aux paramètres RMN de densité protonique et de relaxation, l'expérimentateur dispose d'une panoplie de paramètres d'étude de la structure moléculaire, microscopique et macroscopique. À ces potentialités, il est nécessaire d'ajouter les mesures de température par IRM pour l'étude des transferts thermiques [1], l'étude des phénomènes de transport moléculaire via la mesure des coefficients de diffusion [72] et, enfin, et plus récemment, l'étude des propriétés rhéologiques des matériaux par la mesure de leurs propriétés d'écoulement [73].