

1 Introduction
The catalytic coupling of carbon dioxide and epoxides to afford polycarbonates (Eq. (1)) was first reported by Inoue and coworkers in 1969, employing a heterogeneous catalyst derived from Zn(CH2CH3)2 and H2O [1]. The ubiquitous carbon dioxide molecule provides a low-cost C1 feedstock for production of these commercially applicable polymers via an environmentally benign route. More recently, this process has received a great deal of attention utilizing well-defined, more active, homogeneous catalysts [2–8]. As noted in Eq. (1):

two undesirable reactions may accompany the alternating copolymerization process. That is, successive cyclic ether enchainment leading to polyether linkages, and the one-to-one coupling of CO2/epoxide to produce high-boiling cyclic carbonate byproducts. Hence, part of the efforts in this area have involved understanding ways of controlling these unwanted processes. The most successful catalytic systems to date involve zinc-based complexes, but recently we have published investigations of the mechanistic aspects of the production of polycarbonates utilizing robust five-coordinate Schiff-base chromium(III) initiators similar to those reported by Jacobsen for the asymmetric ring-opening (ARO) of epoxides [9]. These particular complexes were shown to be quite adept at producing polycarbonates from CO2 and the comonomers cyclohexene oxide (CHO) [10,11], propylene oxide (PO) [11,12], and [2-(3,4-epoxycyclohexyl)ethyl]trimethoxysilane [13]. The activity of these complexes and the percentage of copolymer versus polyether is greatly enhanced in the presence of a Lewis base co-catalyst such as N-methylimidzole.
Given the success of the chromium complexes to afford completely alternating copolymer from CO2/cyclohexene oxide with minimal quantities of cyclic carbonate formation, we have undertaken an investigation into the use of similar gallium(III) complexes for the copolymerization of CHO and CO2. Five-coordinate gallium Schiff-base complexes have been previously reported, most notably by Atwood [14,15], which sparked our interest in their potential as copolymerization catalysts due to geometrical similarities to the chromium catalysts and gallium’s electronic resemblance to zinc in terms of d-electron count. In addition, these studies have allowed us to structurally compare analogous metal complexes containing salen ligands with the flexible ethylene vs the more rigid cyclohexyl backbones.
2 Results and discussion
2.1 Synthesis and structure of catalysts
Fig. 1 shows the two Schiff-base ligands used in this study, (cyc)salenH2 (1a) and (et)salenH2 (1b) ((cyc)salenH2 = N,N'-bis-(3,5-di-tert-butylsalicylidene)-1,2-cyclohexyldiimine and (et)salenH2 = N,N′-bis-(3,5-di-tert-butylsalicylidene)-1,2-ethylenediimine). The gallium complexes 2a and 2b were synthesized from the reaction of GaCl3 with the potassium salt of the ligands in THF solution as illustrated in Fig. 2.
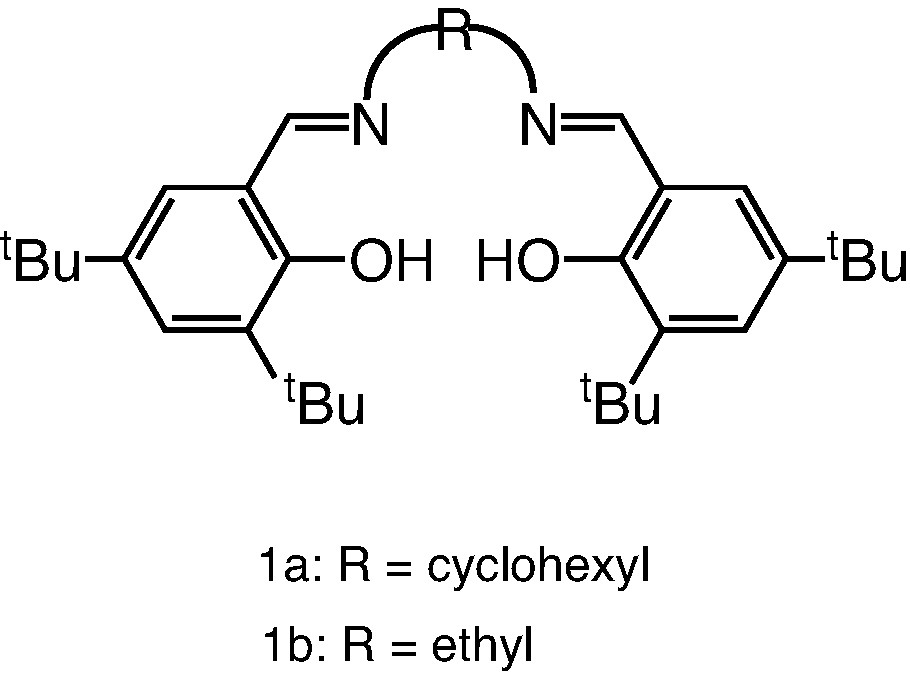
Representation of salenH2.
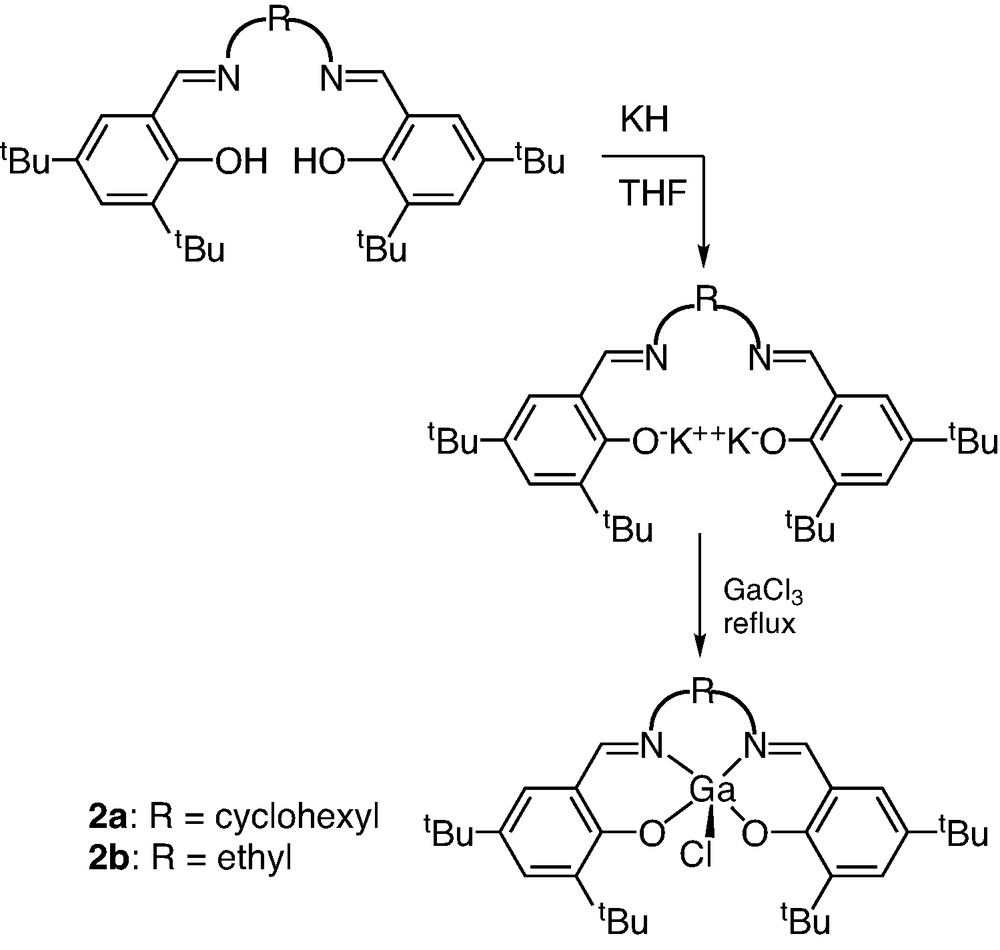
Synthesis of (salen)GaCl.
X-ray quality crystals of 2a and 2b were grown by slow evaporation of a concentrated CH2Cl2 solution of the respective complex into neat toluene. The crystals of 2a are a CH2Cl2 solvate with three disordered solvent molecules present. Crystal and structure refinement data are listed in Table 1.
Crystal data and structure refinement
| 2a | 2b | 3a | 3b | |
| Empirical formula | C39H58Cl7GaN2O2 | C32H46ClGaN2O2 | C36H52GaN5O2 | C32H46GaN5O2 |
| Formula weight | 904.74 | 595.88 | 656.55 | 602.46 |
| Temperature (K) | 110(2) | 110(2) | 110(2) | 110(2) |
| Crystal system | monoclinic | orthorhombic | monoclinic | monoclinic |
| Space group | P2(1)/c | Pccn | P2(1)/c | P2(1)/n |
| a (Å) | 10.281(3) | 15.331(10) | 13.711(4) | 12.970(9) |
| b (Å) | 29.451(10) | 37.57(2) | 23.614(6) | 16.546(11) |
| c (Å) | 14.561(5) | 10.987(7) | 10.639(3) | 15.136(10) |
| B (°) | 91.029(6) | 90 | 98.668(5) | 103.951(13) |
| U (Å3) | 4408(2) | 6328(7) | 3405.2(15) | 3152(4) |
| Dc (Mg m–3) | 1.363 | 1.251 | 1.281 | 1.269 |
| Z | 4 | 8 | 4 | 4 |
| μ (mm–1) | 1.083 | 0.983 | 0.847 | 0.908 |
| Reflections collected | 19831 | 27526 | 15228 | 13944 |
| Independent reflections | 6399 | 4653 | 4912 | 4563 |
| Parameters | 470 | 355 | 409 | 373 |
| Goodness-of-fit on F2 | 1.081 | 0.887 | 1.007 | 1.092 |
| Final R indices [I > 2 σ(I)] | R1 = 0.0984 | R1 = 0.0778 | R1 = 0.0901 | R1 = 0.0441 |
| wR2 = 0.2600 | wR2 = 0.1674 | wR2 = 0.2165 | wR2 = 0.1034 | |
| R indices (all data) | R1 = 0.1468 | R1 = 0.1600 | R1 = 0.1393 | R1 = 0.0711 |
| wR2 = 0.2843 | wR2 = 0.2078 | wR2 = 0.2522 | wR2 = 0.1345 |
A thermal ellipsoid plot of 2a is shown in Fig. 3. The complex possesses slightly distorted square pyramidal geometry. The basal plane has a random mean square (RMS) deviation of 0.0876 from the best-fit plane amongst O(1), O(2), N(1), and N(2) with the gallium ion lying 0.4858(10) Å above the plane. The angles O(2)–Ga(1)–O(1), O(1)–Ga(1)–N(1), and O(1)–Ga(1)–Cl(2) are all very near 90° at 87.5(3)°, 90.5(3)°, and 102.4(2)°, respectively. Further illustration of the distortion is demonstrated by the angles O(2)–Ga(1)–N(1) and O(1)–Ga(1)–N(2), which are fairly obtuse at 144.4(3)° and 155.9(3)°, respectively. The average Ga–O bond distance is 1.867 Å and the average Ga–N distance is 2.016 Å. The Ga–Cl distance of the apical chloride ligand is 2.214(3) Å.

Thermal ellipsoid plot of 2a at 50% confidence level. Solvent molecules and H atoms omitted for clarity.
These distances are in close agreement with those of a similar structure reported by Atwood containing a phenyl backbone as opposed to the cyclohexyl reported here [14]. A thermal ellipsoid plot of 2b is shown in Fig. 4. The structure also displays a greater distortion of square planar geometry with an RMS deviation from planarity by the basal atoms of 0.2755 Å and the Ga atom displaced by 0.4560(32) Å. The bond angles around the basal plane are comparable to 2a with O(1)–Ga(1)–O(2) = 91.7(3)°, O(1)–Ga(1)–N(1) = 89.6(3)°, and O(1)–Ga(1)–Cl(2) = 115.11(19)°. A similar structure was reported by Atwood, in which the apical ligand is an ethyl group as opposed to a chloride [14]. Our average Ga–O distance of 1.877Å is shorter than in Atwood’s structure, while the average Ga–N distance of 2.112 Å is longer. The Ga(1)–Cl(2) distance is 2.213 Å, which is almost identical to that seen in 2a. Fig. 5 depicts an overlay of the coordination spheres in complexes 2a and 2b with the gallium centers fixed in the same position. As is apparent from this representation, there are no significant differences in the structural details of the coordination spheres between salen ligands with the cyclohexyl vs more flexible ethylene backbones.
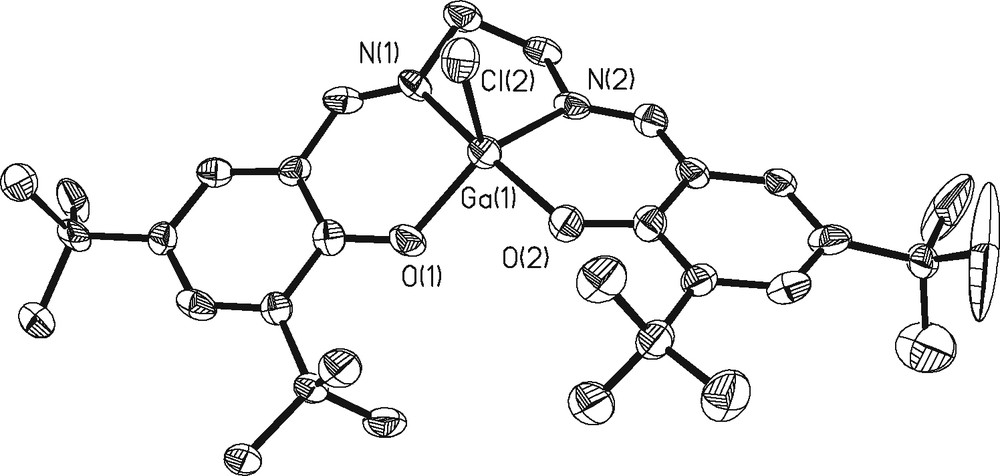
Thermal ellipsoid plot of 2b at 50% confidence level. H atoms omitted for clarity.
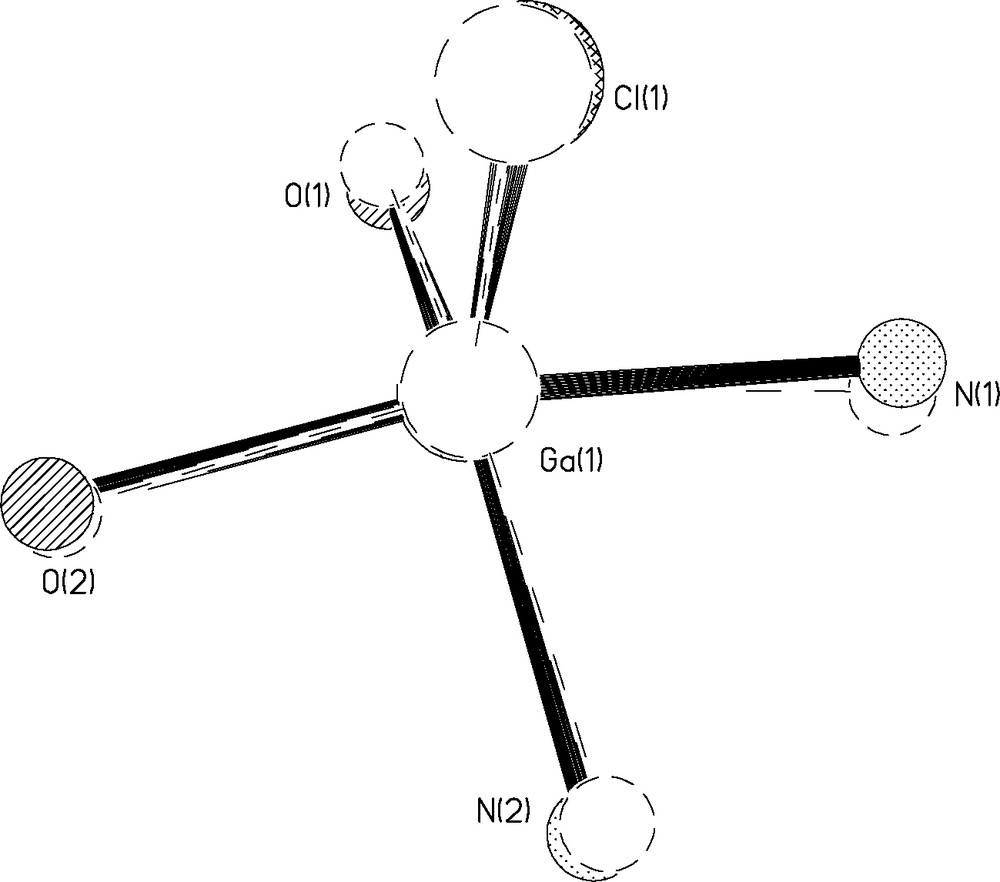
Overlay of the coordination spheres of 2a and 2b. The solid circles correspond to 2a and the dotted circles correspond to 2b.
In order to study the electronic effects of the apical ligand on the catalytic activity of the gallium complexes we replaced the chloride ligand with the more nucleophilic azide group. Complexes 3a and 3b were synthesized from 2a and 2b via reaction with AgBF4 and NaN3, as shown in Fig. 6. The chromium equivalent of 3a has previously been reported by Jacobsen [9], but the synthesis of gallium(III) azide species have not been previously communicated. Unlike the Cr complex, 3a is readily soluble in common non-coordinating solvents such as methylene chloride and toluene, but not in hydrocarbons. A thermal ellipsoid plot of 3a obtained from X-ray quality crystals is displayed in Fig. 7. Again, the five-coordinate gallium structure displays a distorted square pyramidal geometry (RMS deviation = 0.1145Å) with the gallium atom displaced 0.4869(5)Å above the basal plane formed by the coordinated nitrogen atoms N1 and N2, as well as O1 and O2. The Ga-azide bond distance (Ga(1)–N(3)) is 1.918(7)Å and the azide ligand is tilted away from the z-axis (N(4)–N(3)–Ga(1) = 120.0(6)°). Bond distances within the azide are 1.228(10)Å (N(3)–N(4)) and 1.149(10)Å (N(4) – N(5)). Likewise X-ray-quality crystals of 3b were obtained and a thermal ellipsoid plot is given in Fig. 8.
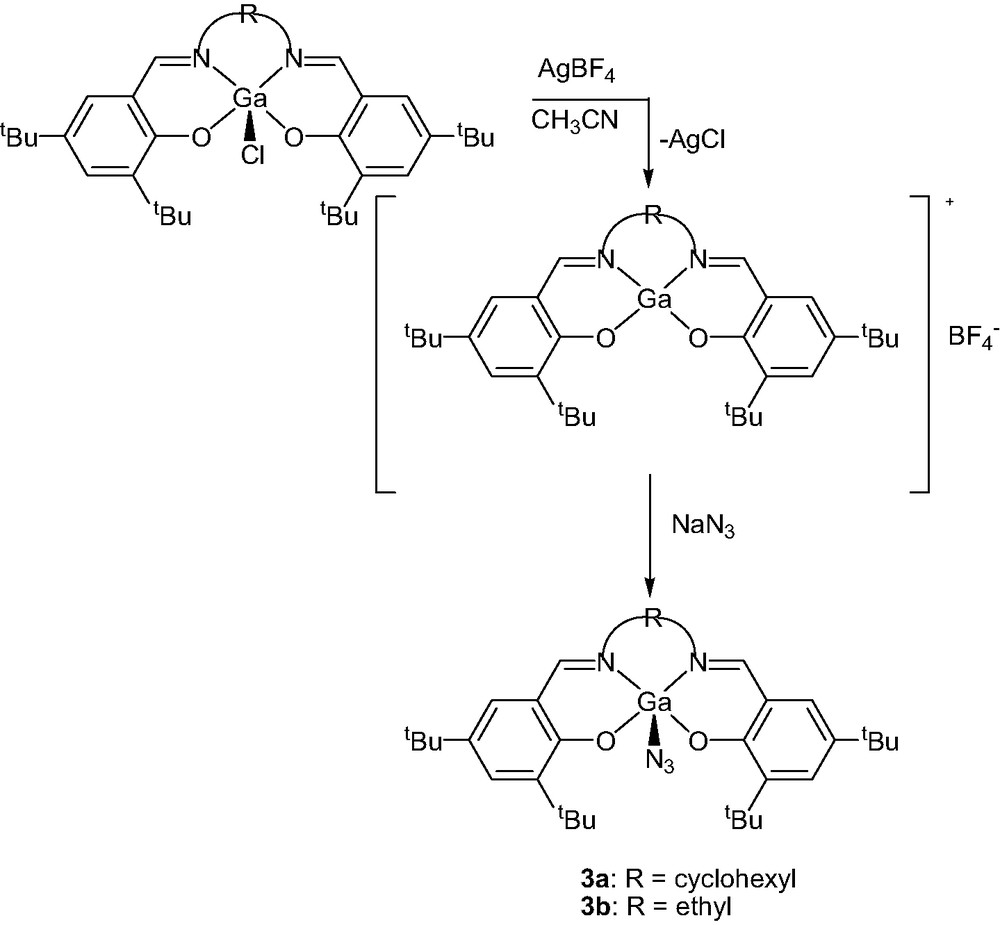
Synthesis of (salen)GaN3 from (salen)GaCl.
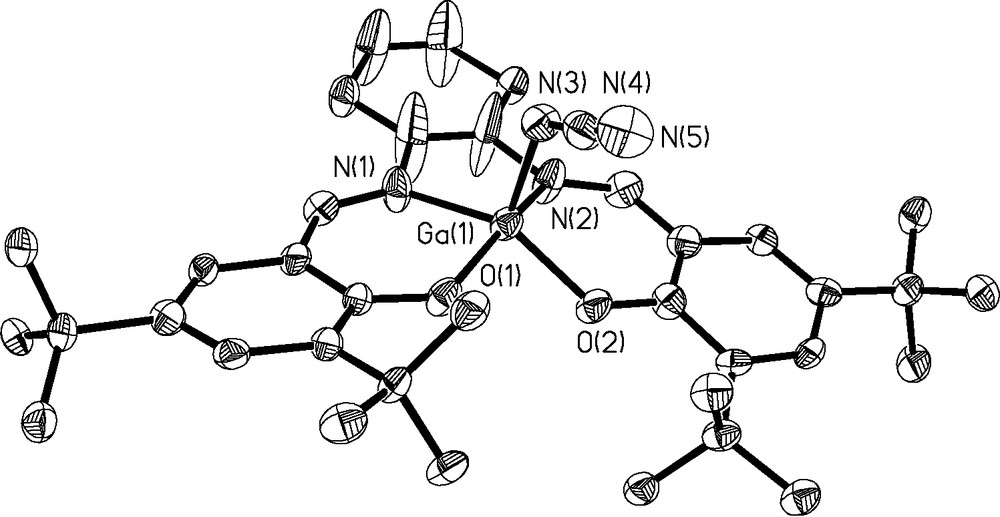
Thermal ellipsoid plot of 3a at 50% confidence level. H atoms omitted for clarity.
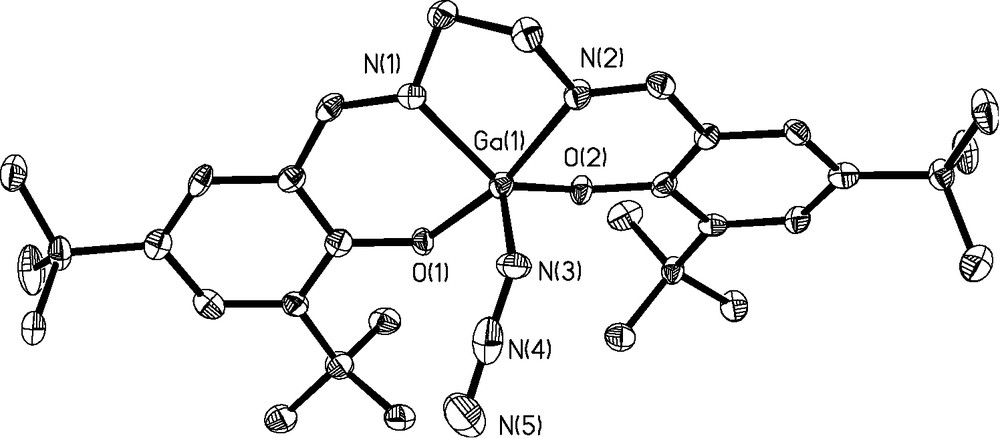
Thermal ellipsoid plot of 3b at 50% confidence level. H atoms omitted for clarity.
As seen in 2b, the increased flexibility of the ethylene backbone over the cyclohexane backbone results in a greater distortion of the square pyramidal geometry with the RMS deviation of N(1),N(2), O(1), and O(2) from the best-fit plane equal to 0.2834Å and the gallium atom lying 0.4985(16)Å above the plane. The Ga–azide bond distance is comparable to that of 3a at 1.897(4)Å with a slightly more pronounced tilt to the coordinated azide ligand (N(4)–N(3)–Ga(1) = 124.0(3)°). Bond distances within the azide are comparable to 3a at 1.203(5)Å (N(3)–N(4) ) and 1.178(6)Å (N(4)–N(5)).
Jacobsen theorizes that the insolubility of the chromium azide complex is due to intermolecular contacts between the azide of one molecule and the open chromium coordination site on a neighboring molecule creating a network that is broken up by coordinating solvent filling the sixth coordination site [9]. It is interesting to note that the unit cell illustrated in Fig. 9 shows no Ga–azide–Ga intermolecular contacts. The molecules, in fact, pack in such a way as to orient the azides away from neighboring molecules.
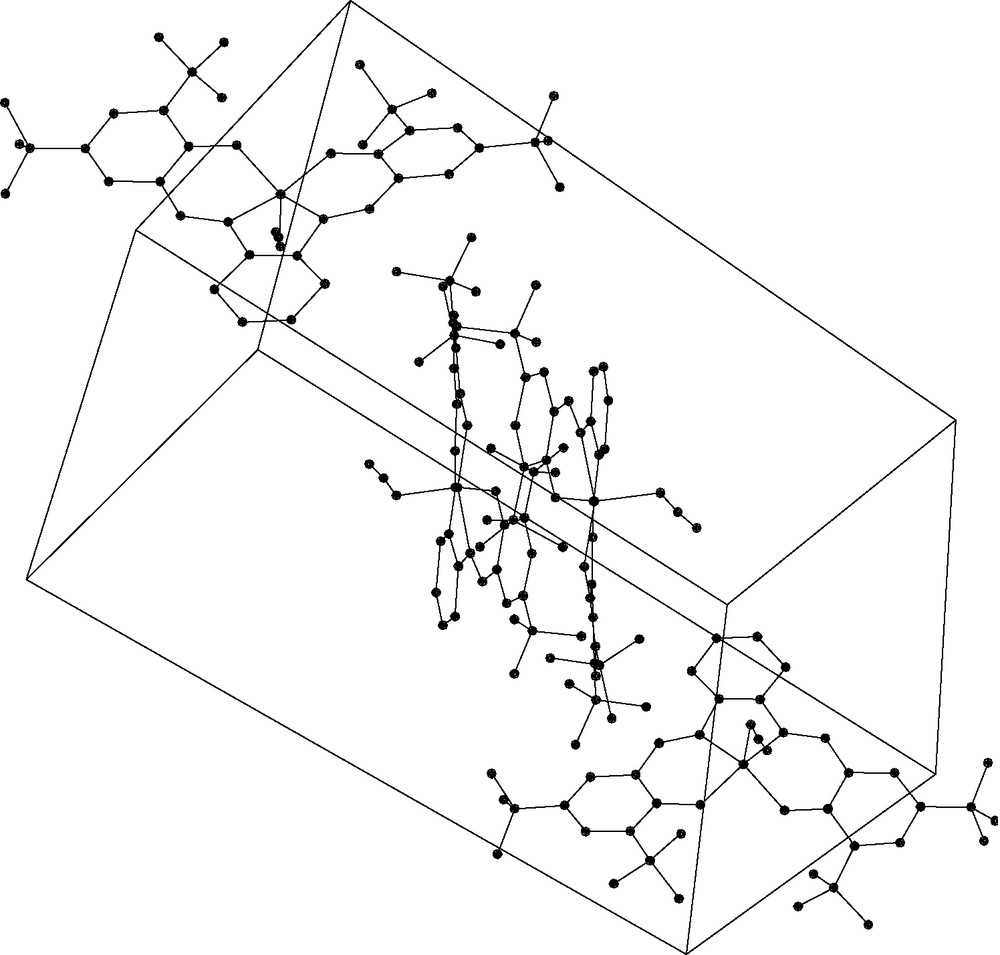
Packing diagram of 3a.
2.2 Epoxide/CO2 copolymerization studies
Copolymerization studies were conducted using 2a and 2b as catalysts for formation of polycarbonates from cyclohexene oxide and CO2. No polymer was observed with either 2a or 2b in the presence of N-methylimidazole. The characteristic infrared stretch of the carbonate found in polycyclohexylcarbonate at ~1750 cm–1 was not observed in the reaction solution. We have previously proposed, in the case of (cyc)salenCr(III)Cl, that the copolymerization is initiated through a second order process in which the nucleophile (Cl) on one chromium center ring-opens a bound epoxide on another chromium center [10,11]. Presumably, in the case of gallium, the imidazole binds to the metal center well enough to preclude displacement by an epoxide even when it is in large excess, and therefore prevents the initiation step. This is not observed in the chromium case as the presence of the Lewis base increases the activity of the catalyst and decreases the percentage of polyether linkages found in the resultant copolymer. Conversely, for an identical copolymerization reaction carried out in the absence of imidazole, a white precipitate forms upon addition of methanol to the reaction solution. A carbonate stretch at 1750 cm–1 was not observed in infrared spectroscopic analysis, which is indicative of the formation of polyether as opposed to polycarbonate. This was confirmed by 1H NMR of the polymer, which shows a broad resonance at 3.6–3.2 ppm, typical of the homopolymer. Concomitantly, no resonance in the region 4.8–4.4 ppm is seen as would be observed in polycyclohexylcarbonate.
An attempt was made to enhance the quantity of copolymer produced by adding only one-half an equivalent of imidazole to the reaction mixture. Presumably, this would leave one-half the Ga(III) complexes available for activating an epoxide. Copolymerization studies with 2b and one-half an equivalent of imidazole produced no observable copolymer. Further attempts were made to enhance copolymer production by replacing the imidazole with a more donating co-catalyst such as triphenylphosphine. Copolymerization studies with 2b and one equivalent of PPh3 as well yielded no observable copolymer.
Polymerization runs conducted utilizing 3a in the absence of a co-catalyst produced no copolymer. In the presence of two equivalents of co-catalyst, in this case a donating phosphine (tricyclohexylphosphine) to further enhance the nucleophilicity of the azide, trace amounts of copolymer were observed in the infrared spectrum of the reaction mixture, but quantifiable amounts of polycarbonate eluded us.
3 Conclusion
We have successfully synthesized two derivatives of (salen)Ga(III)Cl from the potassium salts of the respective ligands. In contrast to similar (salen)Cr(III)Cl imitators, these complexes produced no copolymer from CO2 and cyclohexene oxide in the presence of the Lewis base N-methylimidazole or triphenylphosphine. The complexes produce low molecular weight homopolymer in the absence of a cocatalyst. In addition the azide analogues, (salen)Ga(III)N3, were synthesized. Trace copolymer was observed from CO2/CHO in the presence of tricyclohexylphosphine cocatalyst. Overall the gallium(III) Schiff-base complexes are much poorer catalysts for copolymerization of CO2 and epoxides than their chromium(III) counterparts.
4 Experimental
4.1 Synthesis of (salen)GaCl
A 100-ml Schlenk flask was charged with KH (0.045 g; 1.12 mmol) and stirred in 20 ml THF. To this slurry a 20 ml THF solution of (cyc)salenH2 (0.300 g; 0.55 mmol) was added. The resulting mixture was allowed to stir overnight. A three-necked round bottom flask fitted with a condensor was charged with GaCl3 (0.097 g; 0.55 mmol) in 15 ml of THF. The salen solution was added to the solution of GaCl3 and the resultant solution was refluxed during 6 h, cooled, and filtered. The solvent was removed under reduced pressure and the yellow solid washed with hexanes (2 × 20 ml). Yields 0.286 g (80%) of yellow solid. 1H NMR(C6D6) δ = 1.41 [s, 18H, C(CH3)], 1.92 [s, 18H, C(CH3)], 3.31 [t, 2H, N(CH(CH2CH2CH2)CH)N], 7.08 [t, 2H, Ar–H], 7.60,7.77 [m, 2H, Ar–H], 7.82 [t,2H,Ar–CH]. Elemental analysis: calculated for C: 66.52%; H: 8.06%; N: 4.31%; found: C: 66.34%; H: 8.63%; N: 3.93%.
4.2 Synthesis of (salen)GaN3
A 50-ml Schlenk flask was charged with AgBF4 (0.195 g; 1.0 mmol) and stirred in 10 ml CH3CN. A separate 50-ml Schlenk flask was charged with [(et)salen]GaCl (0.550 g; 0.92 mmol) and dissolved in 30 ml CH3CN. The salen solution was added to the AgBF4 suspension via canula and the resultant mixture was stirred overnight. The mixture was filtered through celite and added to a 20-ml CH3CN suspension of NaN3 (0.065 g; 1.0 mmol). The new mixture was stirred overnight, filtered, and the solvent removed under reduced pressure. Yields 0.542 g (90%) of a yellow solid. 1H NMR (C6D6): δ = 1.37 [s, 18H, C(CH3)], 1.84 [s, 18H, C(CH3)], 2.49 [q, 2H, N(CH2CH2)N], 2.99 [q, 2H, N(CH2CH2)N], 6.81[d, 2H, Ar–H], 7.34[s, 2H, Ar–H], 7.77[d, 2H,Ar–CH] IR(toluene): ν(N3) = 2095 cm–1.
4.3 X-ray diffraction studies
Yellow–green crystals of 2a, 2b, 3a, and 3b were grown by slow evaporation of a concentrated CH2Cl2 solution of the respective complex into neat toluene. Crystals were coated with mineral oil and mounted on a glass fiber at room temperature. A hemisphere of data was collected on a Bruker Smart CCD diffractometer at 100 K [16,17]. Structures were solved via a combination of Patterson maps and direct methods using SHELXS-97 [18]. Solutions were refined utilizing SHELXL as found in the SHELXTL software package [19]. Molecular diagrams and publication materials were produced with programs found in SHELXTL [19]. Crystal and refinement data are listed in Table 1.
4.4 Copolymerizaton studies
0.075 g of catalyst and 2.5 equiv of appropriate co-catalyst (i.e. N-methylimidazole via microsyringe, tricyclohexyl phosphine, triphenyl phosphine) were dissolved in 20 ml cyclohexene oxide and injected via inlet port into a Parr autoclave. The reactor was subsequently charged to 500 psi with bone-dry CO2 and stirred at 80 °C for 24 h. After this time, the CO2 was bled off, the reactor opened, and its contents analyzed via infrared spectroscopy. Methanol was then added to the reactor contents to precipitate any copolymer, which was then analyzed via 1H NMR spectroscopy.