



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Since its discovery over 50 years ago, asymmetric catalysis has enabled the preparation of a wide variety of enantioenriched synthons with different functionalities, with an efficiency in line with green chemistry concepts. A step forward has been taken with the development of heterogeneous asymmetric catalysis, aimed at separating the catalyst from the substrates and reaction products in two different phases (usually a solid and a liquid phase) for easy recovery and reuse of the precious chiral active species. This process has made it possible both to increase the turnover number of the catalysts by recycling them, and to implement flow procedures to best meet industry needs. Kagan and his group pioneered this field by describing the covalent attachment of a chiral rhodium complex analogous to the DIOP ligand to oxidized Merrifield resin and demonstrating its activity in asymmetric hydrogenation and hydrosilylation reactions [1]. They already highlighted the effect of the nature of the support on the activity and enantioselectivity of these reactions, which were subsequently studied and optimized by Stille group [2, 3].
In Kagan’s seminal paper, the rhodium active sites were covalently anchored to the support to prevent leaching of the metal and hence loss of activity, or even contamination of the products. This type of supported catalyst preparation may, however, require several synthesis steps to modify at least the ligand, but sometimes also the support, for strong grafting via the formation of covalent chemical bonds. Interest has therefore been focused on the immobilization of chiral catalysts via non covalent interactions on different supports, with the aim of offering robust systems with simpler preparation but also enabling both the catalyst and/or the released support to be recovered at will, for another use.
Original and long-standing approaches to supported asymmetric catalysis involving the modification of natural chiral supports by metal catalysts should be mentioned here, as did Schwab and Rudolph who studied metal-supported cleaved quartz surfaces [4] or Izumi et al., who used Pd dispersed on silk fibers [5]. This has led to disappointing results in terms of activity and enantioselectivity values, but nevertheless remains a fundamental proof of concept. On the other hand, other procedures were examined in which a conventional metal catalyst was modified by adsorption of natural chiral molecules. This is the case, for example, with highly efficient hydrogenation catalysts such as tartaric acid-modified Ni catalysts or cinchona-modified Pt catalysts. In the presence of such metallic surfaces modified by these chiral inducers, the asymmetric transformation is due to the selective adsorption of the substrate onto the chiral surface, leading to very convincing results, for the hydrogenation of β-ketoesters or β-diketones for the first catalyst, and for the hydrogenation of α-ketoesters for the second one. This field of research has been the subject of numerous reports, including a comprehensive review by Blaser [6], and these methods have also been successfully applied on a large scale, for example in the preparation of Benazepril, an angiotensin-converting enzyme inhibitor intermediate [7].
However, major difficulties have arisen in achieving reproducible reaction rates and optical yields, as the selectivity of the transformations is highly dependent on the catalyst preparation procedure and/or its modification with the chiral inducer. Enantioselectivity indeed results from specific interactions between the substrate, the chiral modifier and the metal, the nature of which remains largely unknown and uncontrolled. Work in this field has now mostly shifted to the use of well-defined chiral active catalysts whose activity is more predictable and reproducible thanks to their immobilization on different supports [8, 9, 10, 11, 12, 13, 14].
Non-covalent immobilization of enantioselective catalysts was described in exhaustive reviews nearly 15 years ago, concerning organometallic [15] or organic catalysts [16], organized according to the immobilization method used. At that time, most of them involved electrostatic, acid-base, hydrophobic or coordination interactions, adsorption or some entrapment methods. Since then, new strategies have emerged involving π-stacking or charge-transfer interactions, coming from our group and others. Accordingly, this account will highlight some of the most recent results describing non-covalent interactions for the easy recovery and reuse of enantioselective catalysts, starting with a selection of articles dealing with the use of ionic interactions with inorganic supports to immobilize catalysts. Then asymmetric catalysis with active chiral species immobilized by π-stacking interactions will be summarized, as well as examples implying the formation of charge transfer complexes. Finally, we have chosen to present some of the most recent examples of coordination interactions, such as those leading to metal organic frameworks (MOFs), as they also enable recent, contextual and valuable asymmetric multicatalysis. The vast majority of the examples we have chosen to highlight here involve the use of the well-known salen complexes, due to their ease of access and their high efficiency in a very large number of asymmetric catalysis reactions.
2. Electrostatic interactions with inorganic supports
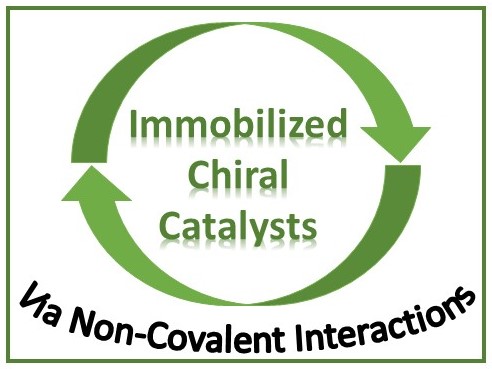
As previously mentioned, the non-covalent immobilization of chiral catalysts has already been widely developed and mainly involved anchoring complexes on silica supports, modified accordingly to allow predominantly ionic interactions via sulfonic acid bonds. As an example, Kim and coworkers have described the hydrothermal synthesis of sulfonic acid-functionalized SBA-16 mesoporous silica [17]. The incorporation of active Co-salen moieties was achieved by the addition of Co(II) complexes in solution and subsequent oxidation in air leading to a stable linkage in the form of ionic pairs with sulfonate anions on the surface. The robustness of the solid catalyst was tested for the Asymmetric Ring Opening (ARO) of rac-epichlorohydrin by water or phenol derivatives. The reactions even gave better results than those carried out with analogous homogeneous catalysts, i.e. with lower catalyst loading and shorter reaction time. The targeted products were recovered, after simple solid filtration, with enantioselectivity values exceeding 98% in some cases and yields reaching 45% (Scheme 1). A slight drop in activity was observed when recycling was attempted in the hydrolytic kinetic resolution of rac-epichlorohydrin, leading to a polar diol, which may compromise ionic pairing on silica. In the case of the formation of less polar compounds, using phenols as nucleophiles for example, no loss of activity was observed and the catalyst could be reused. The authors proved that the support could be recovered and reused after a new Co-salen re-attachment; they also demonstrated that no reaction occurred in the supernatant after catalyst removal before complete conversion, as a proof of the effectiveness of the heterogeneous procedure.
Immobilization by ion pair interactions for the ARO of epoxides.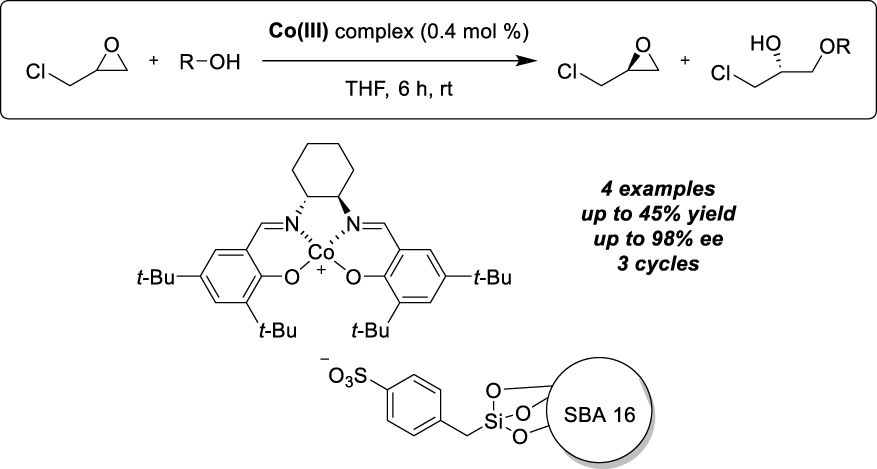
New developments include the use of a different support, namely chiral silica, in the hope of synergy for possible matching effects between the two chiral elements, the support and the complex. In this context, Sun and his group prepared a series of helical silica materials in a sol–gel procedure doped with sodium lactate to affect chirality, and carried out subsequent phenylsulfonylation before immobilizing Mn-salen complexes in ionic pairs [18]. Comprehensive analytical studies were performed to prove the immobilization of the chiral catalysts and the integrity of the helical structure. These new structures were tested as catalysts to promote the hetero-Diels–Alder reaction between Danishefsky’s diene and various aldehydes. Active and enantioselective catalysts were obtained after immobilization of achiral Mn-complexes, demonstrating the chiral induction induced by the support alone. Immobilization of a chiral catalyst improved enantioselectivity values, the molecular catalyst being responsible for product configuration and the chiral environment for enantioselectivity enhancement. The authors describe a series of four different aldehydes and the reaction with acetophenone with varying success, which they attribute to restricted conformational changes by hydrogen bond or other adsorptions on the support, depending on the substrate structure. The transformation of heptaldehyde, however, has been described with total conversion and selectivity (Scheme 2). A major influence of solvents was observed, with ionic liquids influencing the product configuration and promoting catalyst recyclability. Six cycles of the solid catalyst in BMImBF4 gave stable results in terms of product yield (74%) and enantioselectivity (>99%). These chiral helical silica supports have also been studied for their ability to promote asymmetric epoxidations of non-functionalized alkenes, in the presence of PhIO or m-CPBA [19]. In this case the silica support has been modified with pendant ammonium groups and the Mn-salen derivative was modified on the aryl moieties by a sulfonyl group, remote from the catalytic center, to enable ion pair formation. Again, an enantioselective transformation occurred in the presence of the exclusive helical chirality, and synergistic effects were highlighted between the mesoscopic and the molecular chirality, with match or mis-match properties.
Immobilization by ion pair interactions on chiral silica for the hetero Diels–Alder reaction.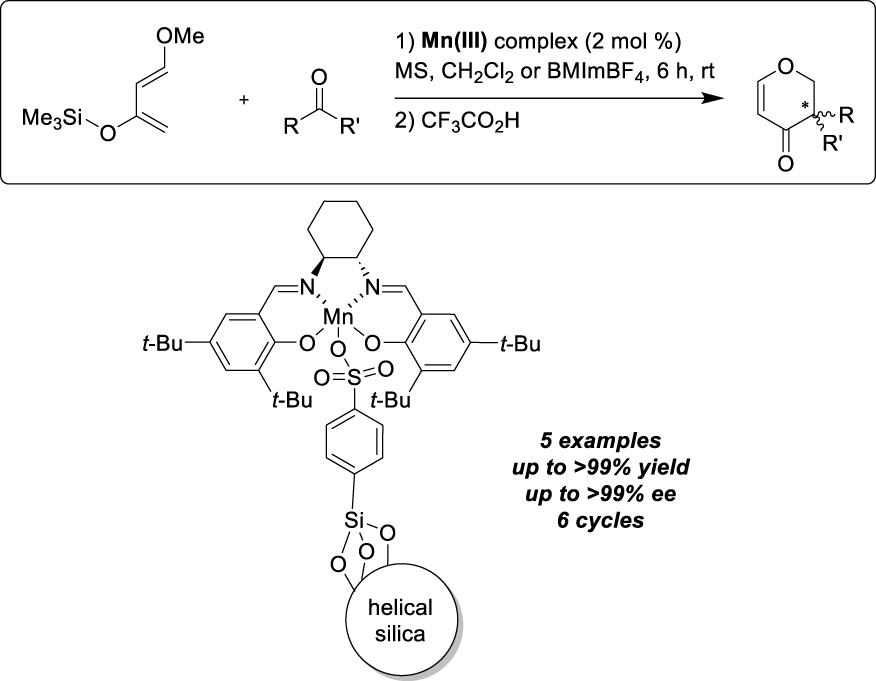
Other types of supports, namely layered zinc or Lanthanum Hydroxide Nitrates (LHS, Layered Hydroxide Salts) have also been used as ion-exchangeable supports to immobilize Mn-salen complexes modified with sodium sulfonate groups [20]. The lower inter-layer forces allowed indeed the intercalation of larger anions, even though FT-IR analyses revealed only partial exchange of nitrates by the anionic catalyst. The benchmark reaction involved the enantioselective epoxidation of styrene derivatives and the Zn-LHS-supported Mn-salen sandwich structure gave the best results in terms of activity and selectivity, achieving 79% conversion and 99% ee for the epoxidation of α-methylstyrene with iodosylbenzene (Scheme 3). Recycling can be achieved after simple filtration and the epoxidation was successful for the following four cycles.
Layered Hydroxide Salts as ion-exchangeable supports for alkenes epoxidation.
Ionic links of different nature have further been exploited towards immobilization of metal-salen derivatives on silica supports. Bhaumik, Islam et al. prepared a chemically transformed carboxylic acid functionalized mesoporous silica (AFS-1) by modification of the SBA-15 outer sphere [21]. A methylammonium-modified Co-salen complex was then immobilized by ionic exchange interactions between the anionic support and the cationic complex. Asymmetric aminolysis of racemic and meso epoxides (10 examples) has been achieved with excellent results under solvent-free conditions, leading for example to the formation of 2-(phenylamino)phenol in 97% isolated yield and >99% ee. The high recyclability of the catalyst has been described, as it can be engaged in successive transformations up to 5 times with almost no loss of efficiency (Scheme 4A). An analogous catalytic material was also prepared with the corresponding Cu-complex and found to promote the asymmetric nitroaldol reaction between various aryl- or alkylaldehydes and nitromethane (11 examples) with high enantioselectivity (up to 94%) and TONs [22]. The recyclability of the procedure was demonstrated by performing the multiple reaction of p-nitrobenzaldehyde with nitromethane with the same batch of catalyst in dichloromethane, which proceeded successfully with only a slight loss in activity (95% to 84% conversion) and an identical enantioselectivity value (91%) for eight consecutive cycles (Scheme 4B). Interestingly, this supported catalyst has been used to prepare (R)-isoproterenol, a non-selective β-adrenoreceptor agonist.
Immobilization by ion pair interactions on mesoporous silica for aminolysis of epoxides and nitroaldol reactions.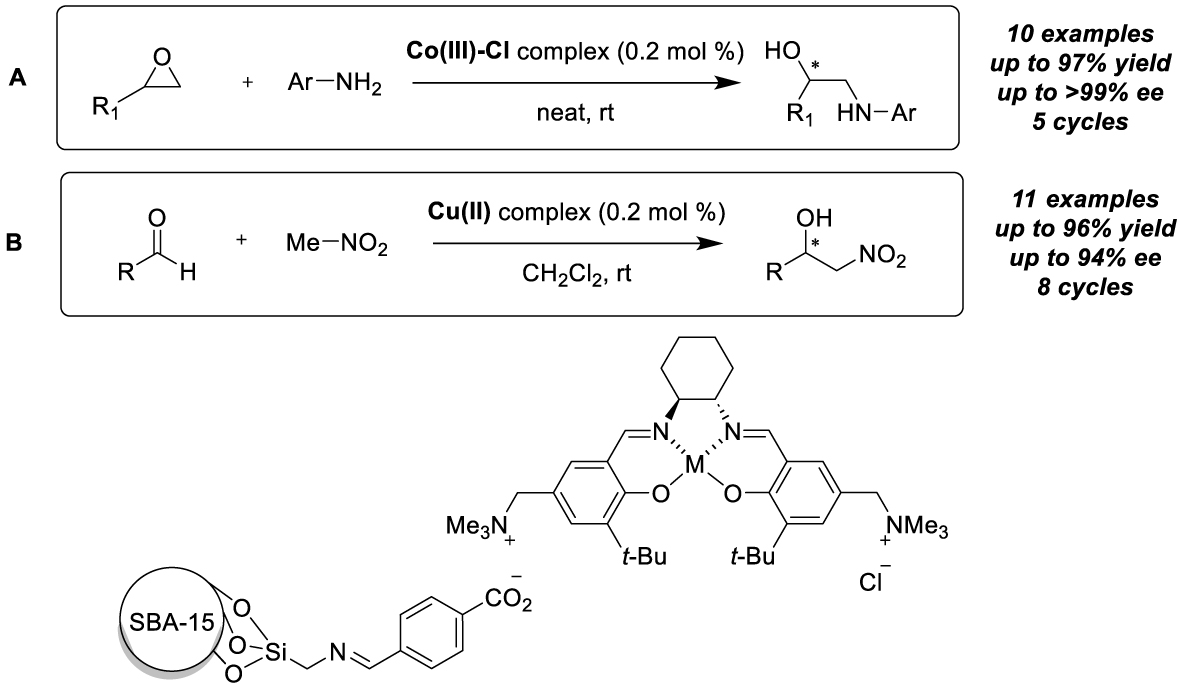
The last example we have chosen to present concerns the preparation of heterogeneous Mn-salen complexes anchored on mesoporous materials functionalized with imidazole groups [23]. Modified silica supports were obtained by co-condensation of tetraethyl orthosilicate and N-(3-triethoxysilylpropyl) imidazole, resulting in functionalized MCM-41, favoring homogeneous distribution of organic species. Immobilization of the Mn-salen complex was achieved in refluxing toluene to form electrostatic bonds with the imidazole group, resulting in a solid catalyst offering high activity and enantioselectivity similar to that obtained with soluble catalysts, in the epoxidation of olefins (3 examples, up to 99% conversion and 85% ee for the oxidation of indene) (Scheme 5). It should be noted that very small quantities of catalyst could be used, due both to the high electron-donating capacity of the imidazole ligand and the good accessibility of the catalytic sites. Virtually no metal leaching was detected by ICP-AES analysis (Inductively Coupled Plasma Atomic Emission Spectrometry); nevertheless, the supported catalyst progressively lost its efficiency when subjected to recycling experiments (3 runs).
Immobilization by ion pair interactions on MCM-41 for alkenes epoxidation.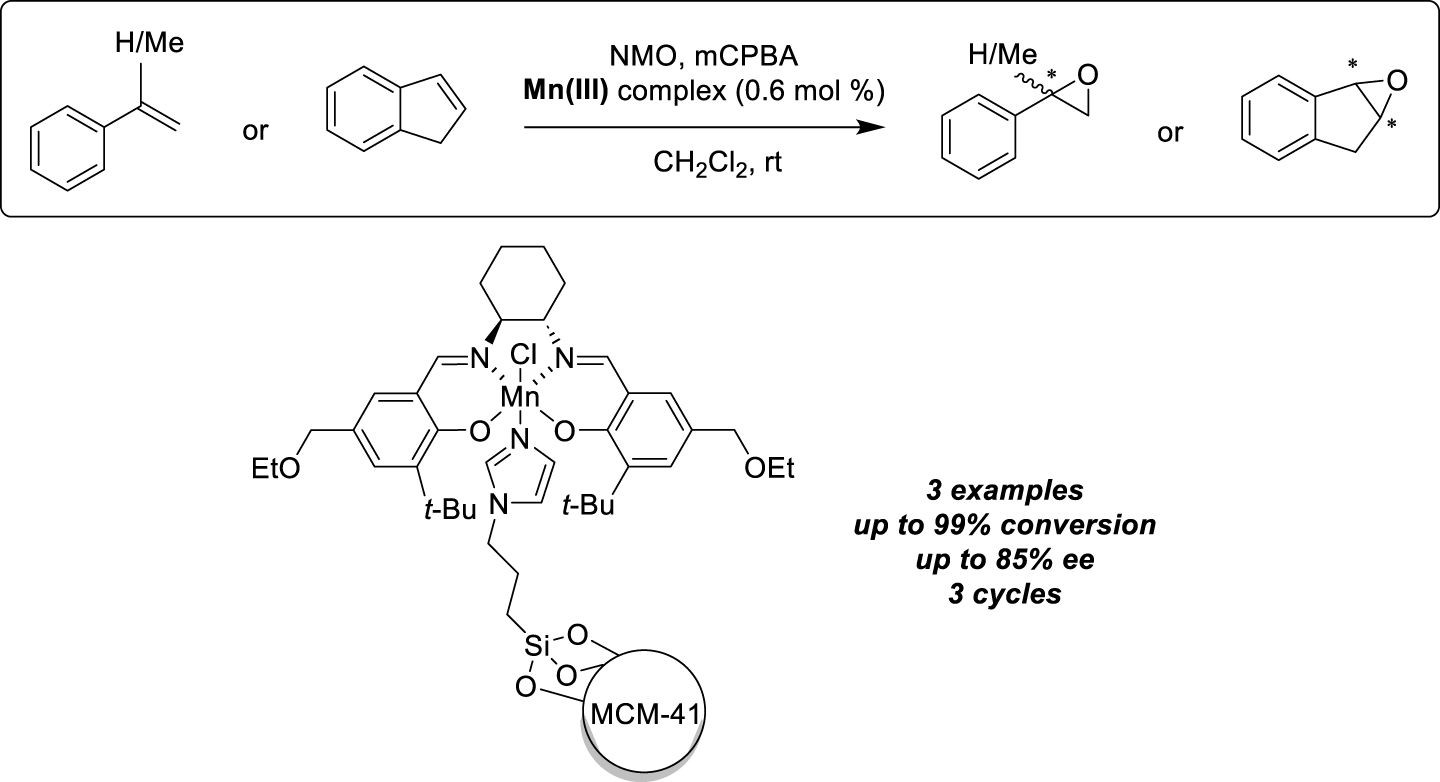
Other types of electrostatic interaction than those reported previously are existing, namely hydrogen bonding which has been used, however not specifically for the immobilization of catalysts on a solid support, but to bring two salen complexes together, with the aim of promoting their cooperativity [24]. Hong and his group have prepared unsymmetrical Co-salen complexes bearing complementary 2-pyridone/aminopyridine moieties on each phenolic group, capable of creating self-assembled dimers in solution via hydrogen bonding. The reaction tested was the Henry reaction, in which both the aldehyde and the nitronate ion are activated by the cobalt center so that the asymmetric catalysis can take place in the presence of a tertiary amine, as a base. Control experiments revealed significant rate acceleration using this assembled catalyst, as well as kinetic studies indicated a second order relative to the cobalt concentration, with excellent enantioselectivity values for a range of aldehydes (8 examples, up to 96% ee, Scheme 6A). These authors demonstrated that other functionalities, notably the urea unit, could be used to create directional hydrogen-bonding interactions for self-assembly [25]. Therefore, they prepared various symmetrical Co-salen complexes functionalized with bis-urea units and studied their efficiency in catalyzing the HKR (Hydrolytic Kinetic Resolution) of rac-epichlorohydrin; these dimeric species (11 examples) were found to exhibit a significant rate acceleration compared with the monomeric catalyst, both in solution (in THF, up to 92% ee) and under solvent-free conditions (up to 93% ee), and this proved true for the transformation of four different terminal epoxides (Scheme 6B). In this case, kinetic studies also showed that the rate laws were second-order in the cobalt concentration and further mechanistic studies were carried out, consistent with the fact that the observed rate enhancement was due to the self-assembly by urea-urea hydrogen bonding.
Immobilization by hydrogen bonding for nitroaldol reactions and hydrolysis of epoxides.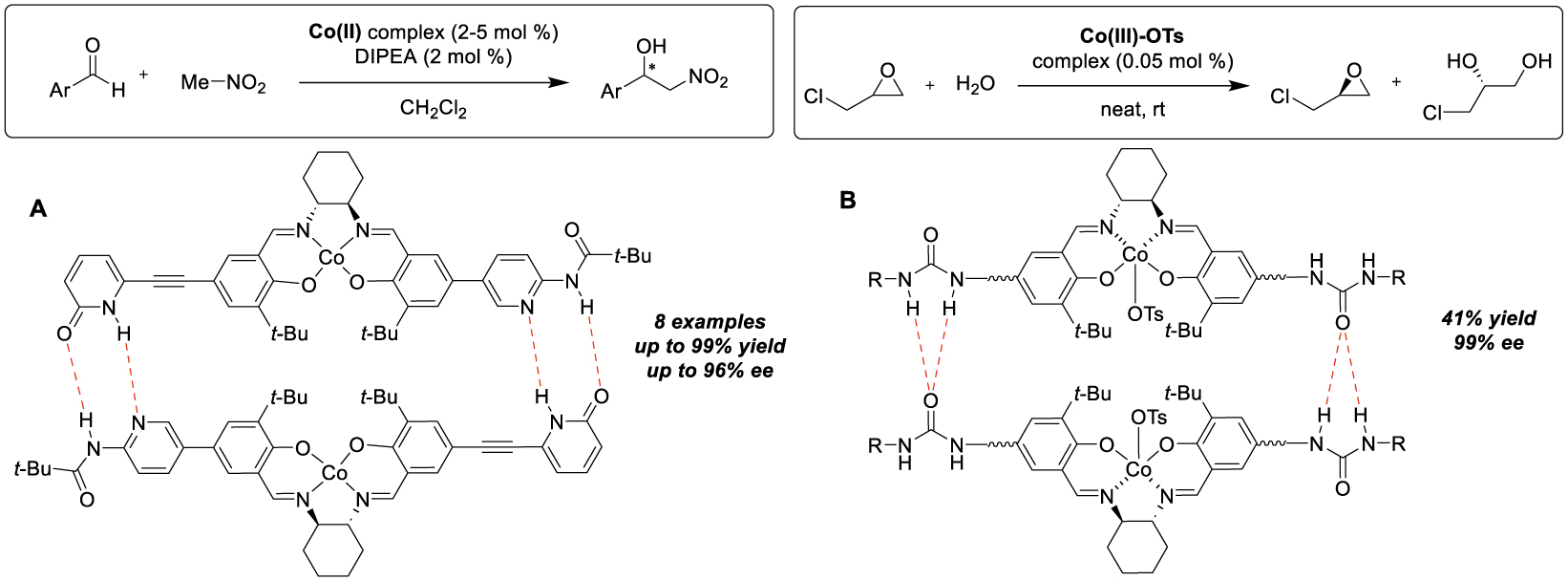
3. π-stacking interactions with carbon supports
In 2008, Zhou et al. reported an example of enantioselective hydrogenation of α-dehydroamino esters catalysed by a Rh(I) complex immobilized via a pyrene-modified PyrPhos ligand on Carbon Nanotubes (CNTs) [26]. This ligand is readily accessible by simple amide coupling, and both the activity and enantioselectivity of the corresponding catalyst are unaffected by the presence of the pyrene-tag. Various examples of substrates containing electron-donating or electron-withdrawing para-substituted aromatic groups have led to the targeted products in excellent yields (>99%) and very good enantiomeric excess values (92–96% ee). The main advantage of these modified catalysts remains their solvent-dependent adsorption capacity. As such, reactions can be carried out under homogeneous conditions, and the catalyst recovered by filtration after a simple change of solvent for precipitation. The authors showed that the lowest adsorption rate on CNTs was obtained when using dichloromethane (50%), which was considered the best solvent because the catalyst was fairly mobile and the active sites easily accessible. On the contrary, the use of a solvent with a high adsorption rate on CNT, as ethyl acetate, led to the precipitation of the complex, making it suitable for catalyst recovery and recycling by filtration. By playing on this difference of adsorption, easy catalysts recycling was performed by a simple solvent change and 9 cycles were realized without affecting either the conversion or the enantioselectivity (Scheme 7A). A similar transformation was reported by Fernández, Khiar and their group in 2017, for which they proposed a pyrene-tagged carbohydrate-based phosphorus/sulfur ligand for Rh(I) catalysis immobilized on Single-Walled Carbon Nanotubes (SWCNTs) [27]. Here, the effect of carbon linker length on catalytic activity was investigated for the hydrogenation of methyl α-acetoamido-cinnamate. Notably, they demonstrated low activity using a one-carbon linker, whereas a three-carbon linker achieved the same conversion and enantioselectivity as with a traditional Rh complex in dichloromethane (>99% conversion, 90% ee). In addition, they also showed that the presence of this pyrene-tag made the reaction efficiency more solvent-dependent, probably due to possible competitive complexation of Rh(I) with the aromatic rings to form Rh(I)-η6-arene complexes. As mentioned previously [26], the solvent also influenced the catalyst adsorption on CNTs, allowing reactions to be carried out under homogeneous conditions and the catalyst to be recovered by changing the solvent. Here also, dichloromethane was the solvent of choice to carry out the reaction. On the other hand, while the reaction went poorly in ethyl acetate, the use of this solvent enabled almost complete adsorption onto the CNTs after one hour of sonication, which allowed to switch to heterogeneous conditions for easy catalyst recovery. Based on these results, four successive runs were carried out with the same batch of catalyst, starting with excellent conversion (>99%) and enantiomeric excess (96%) to dramatically decreased values at the end (<10% conversion, 40% ee), probably due to the intrinsic instability of the catalyst (Scheme 7B). Later, in 2020, Shi et al. described a method for the asymmetric hydrogenation of dehydroamino acid esters under heterogeneous conditions [28]. They employed pyrene-modified MonoPhos-Rh(I) complexes immobilized on graphene, demonstrating both high catalytic activity and enantioselectivity. The use of ethyl acetate enabled the achievement of comparable catalytic performance to classical homogeneous conditions, coupled with high adsorption on graphene. The study encompassed eight examples of substrates featuring variously substituted aromatic groups. This resulted quantitative yields and enantiomeric excess values ranging from 91 to 96% for the targeted products (Scheme 7C). Notably, diverse analytical methods were employed to confirm well-defined immobilisation. 31P Cross-polarization magic-angle spinning (CP-MAS) NMR spectra demonstrated that the Rh complexation pattern remained unchanged after adsorption on graphene surfaces with a 3 × 10−4 mmol⋅mg−1 loading on graphene. Additionally, EDS TEM analyses (Energy Dispersive Spectroscopy for elemental mapping in Transmission Electron Microscopy) illustrated a homogeneous distribution of the Rh complex on the surface. BET (Brunauer–Emmett–Teller) measurements indicated a surface area for the immobilized catalyst reduced by half compared to the raw graphene. The catalyst exhibited reusability for up to 13 cycles without compromising the conversion of the starting material or enantiomeric excess values. However, it is worth mentioning that the reaction time had to be extended from 2 h to 10 h after seven cycles, likely due to the instability of the filtrated intermediate in the absence of a hydrogen atmosphere.
π-stacking interactions on CNTs for Rh-catalyzed asymmetric hydrogenation.
Another example of immobilised Rh(I)-catalysed reaction on carbon surfaces has been developed by Godard and his group in 2019 [29]. The authors focused on the asymmetric hydroformylation of norbornene to preferentially obtain the exo-product, utilizing new pyrene-tagged diphosphite ligands supported on various carbon materials and adapted for continuous flow conditions (Scheme 8). Initially, the authors designed new sugar-based ligands to achieve good exo-product yields (>99%) and enantiomeric excess values (62%) under homogeneous conditions. Optimization efforts revealed that glucofuranose-diphosphite ligands exhibited better enantioselectivity than xylofuranose-derivatives, especially when introducing substituents on the biphenyl moiety. Moreover, the O-alkylation at the glucofuranose-C5-position played a crucial role in catalyst activity. Subsequently, the heterogenization of the reaction was undertaken by adding a pyrene-tag to the effective ligand, ensuring no modification of the catalyst activity. The immobilization of the tagged Rh(I)-complexes was studied on different carbon materials (Multi-Walled Carbon NanoTubes (MWCNTs), Reduced Graphene Oxide (rGO) and Carbon Black (CB)). The results were consistent when using MWCNTs or rGO but significantly increased on CBs, likely due to the entrapment of the complexes in the material structure. The effect of carbon-linker length was highlighted, with the four-carbon linker on MWCNT showing the best results being selected for the rest of the study. However, heterogeneous catalysis resulted in significantly lower enantioselectivity values, and in terms of the recycling ability of the immobilized catalyst in batch conditions, no conversion of the starting material was observed after three runs. ICP-MS (Inductively Coupled Plasma Mass Spectrometry) analysis demonstrated high Rh leaching during the reaction. Nevertheless, with the most optimized conditions in hand, the authors have tested continuous flow conditions. Injecting the syngas mixture via a mass flow controller, they optimized the reaction at 10 bar pressure to achieve higher enantioselectivity in the flow mode (72%) than in batch (62%). This improvement was attributed to more efficient gas-mass transfer which increased gas solubility, and better exposure of substrates to the catalyst, resulting in a higher local catalyst concentration. Subsequently, the support was changed for rGO or CBs, and similar enantioselectivity was obtained with approximately the same substrate conversion and lower decay. This was explained by a larger surface area of rGO (450 m2⋅g−1) and CBs (211 m2⋅g−1) compared to MWCNTs (80 m2⋅g−1) and improved ligand interactions with those supports, facilitated by the presence of functional groups on rGO or entrapment in mesoporous CBs. Although increasing the catalyst loading in CBs support allowed to increase the conversion up to 35% with the same enantiomeric excess value, Rh leaching remained a significant drawback for this reaction.
π-stacking interactions on CNTs for Rh-catalyzed asymmetric hydrogenation of norbornene.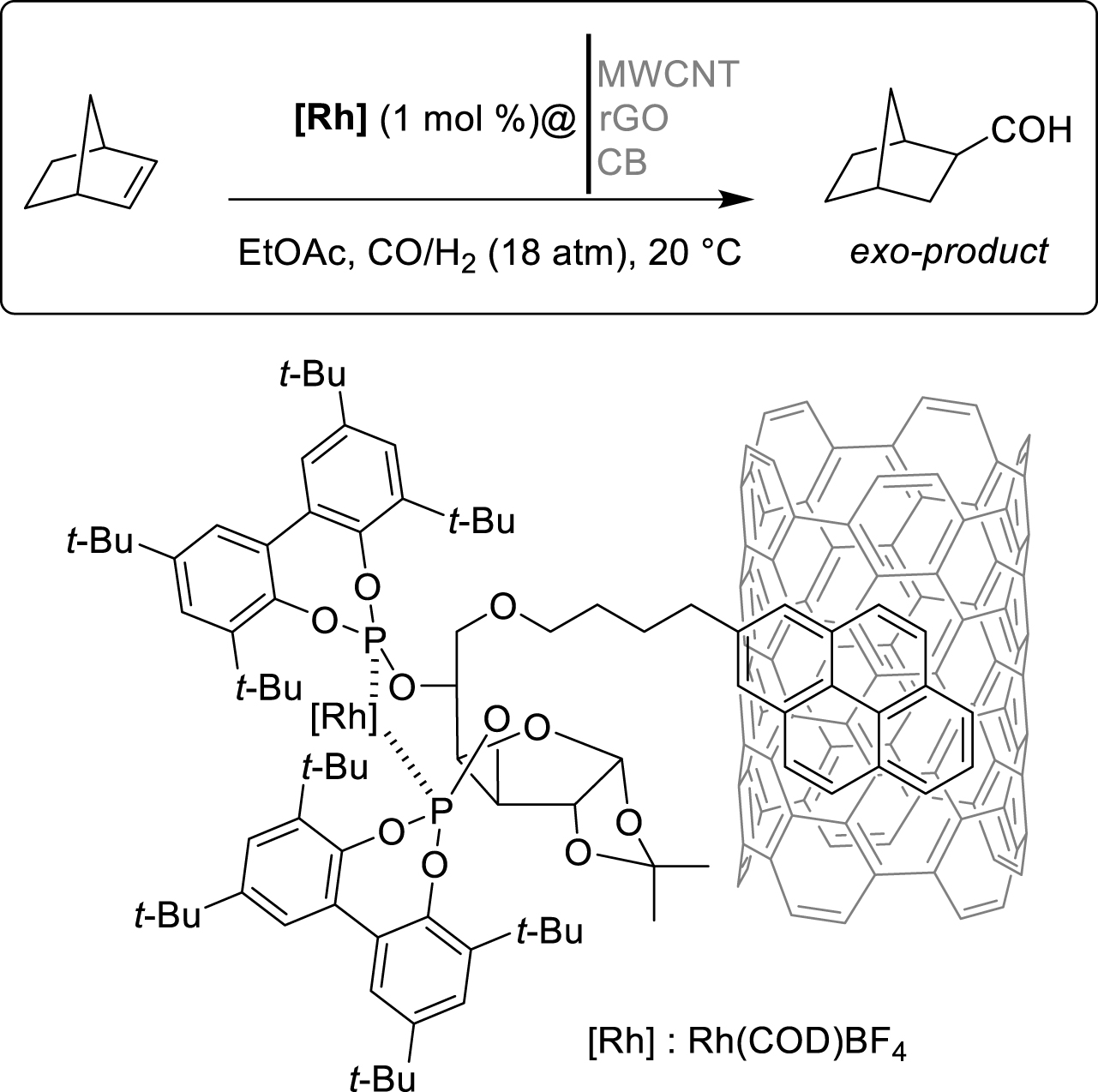
In 2013, Schulz et al. described the use of immobilized bis(oxazoline)-ligands for both copper-catalyzed Henry and ene reactions (Scheme 9) [30]. The use of anthracene-tagged ligands was favored to optimize the reaction conditions and study the recyclability of the supported catalysts. First, the Henry reaction between benzaldehyde and nitromethane was carried out using an anthracene-bis(oxazoline)-tagged Cu(OAc)2 complex in ethanol under homogeneous conditions, which furnished excellent yield (89%) and enantiomeric excess (90%). However, after immobilization on activated carbon, the yield and enantioselective excess value slightly decreased (68% isolated yield, 69% ee), indicating that the support interferes with the reaction, leading to less accessible active sites. Moreover, formation of traces of elimination by-products was observed, probably due to the non-negligible acidity of the support, explaining the loss of yield. Then, recycling of the catalyst was achieved after centrifugation of the reaction medium to recover the catalyst. After 6 runs, the reaction time increased from 24 to 120 h for moderate yields and enantiomeric excesses. A test reaction performed with the supernatant after a recycling cycle gave comparable results, showing that a non-negligible amount of catalyst had been solubilized during the washing. Then, the reaction scope was extended to the ene reaction between ethyl glyoxylate and 𝛼-methyl-styrene with the anthracene-bis(oxazoline)-tagged Cu(OTf)2 catalyst in dichloromethane. In this case, the same activity and enantioselectivity were obtained compared with homogeneous conditions (89% yield, 67% ee). However, throughout the runs, a slight decrease in activity and enantiomeric excesses was observed, as in the previous case. The catalyst immobilization on charcoal was then experimented and gave similar good results (85% yield, 70% ee). Attempts to recover and reuse the free ligand, exempt of the anthracene-tag, gave poor results during the recycling, indicating that those tags are really necessary for the catalyst recovery, without affecting its activity. Finally, to diversify supports, studies using a pyrene-tag instead of an anthracene one has been conducted to compare the effect of various carbon surfaces: charcoal, fullerene and SWCNT. After one run, some differences were already observable: all supported Cu(OTf)2-supported catalysts allowed to obtain the desired product of the ene reaction with excellent yields (89%) but with variable enantioselectivity. The best enantiomeric excess was obtained with the catalyst supported on charcoal (69% ee), followed by SWNCT-supported reaction (64% ee) and then fullerene (56% ee). Pyrene- or anthracene-tags showed no change in recyclability, despite a stronger π–π interaction in the former case. After seven runs, the charcoal-supported catalyst showed a slight decrease in yield and enantiomeric excesses. For the fullerene-based catalyst, the values remained more stable due to strong stacking with the pyrene-tag, which allowed easy recyclability throughout the cycles, but with lower enantioselectivity. On the other hand, the SWCNT-supported Cu(OTf)2 complex gave better results in terms of activity, and values remained constant. This support seemed to be the best immobilization method, avoiding leaching, and promoting strong interactions with the pyrene-tag to ensure a good anchoring of the catalyst for recycling.
π-stacking interactions on carbon supports for Cu-catalyzed nitroaldol and ene reactions.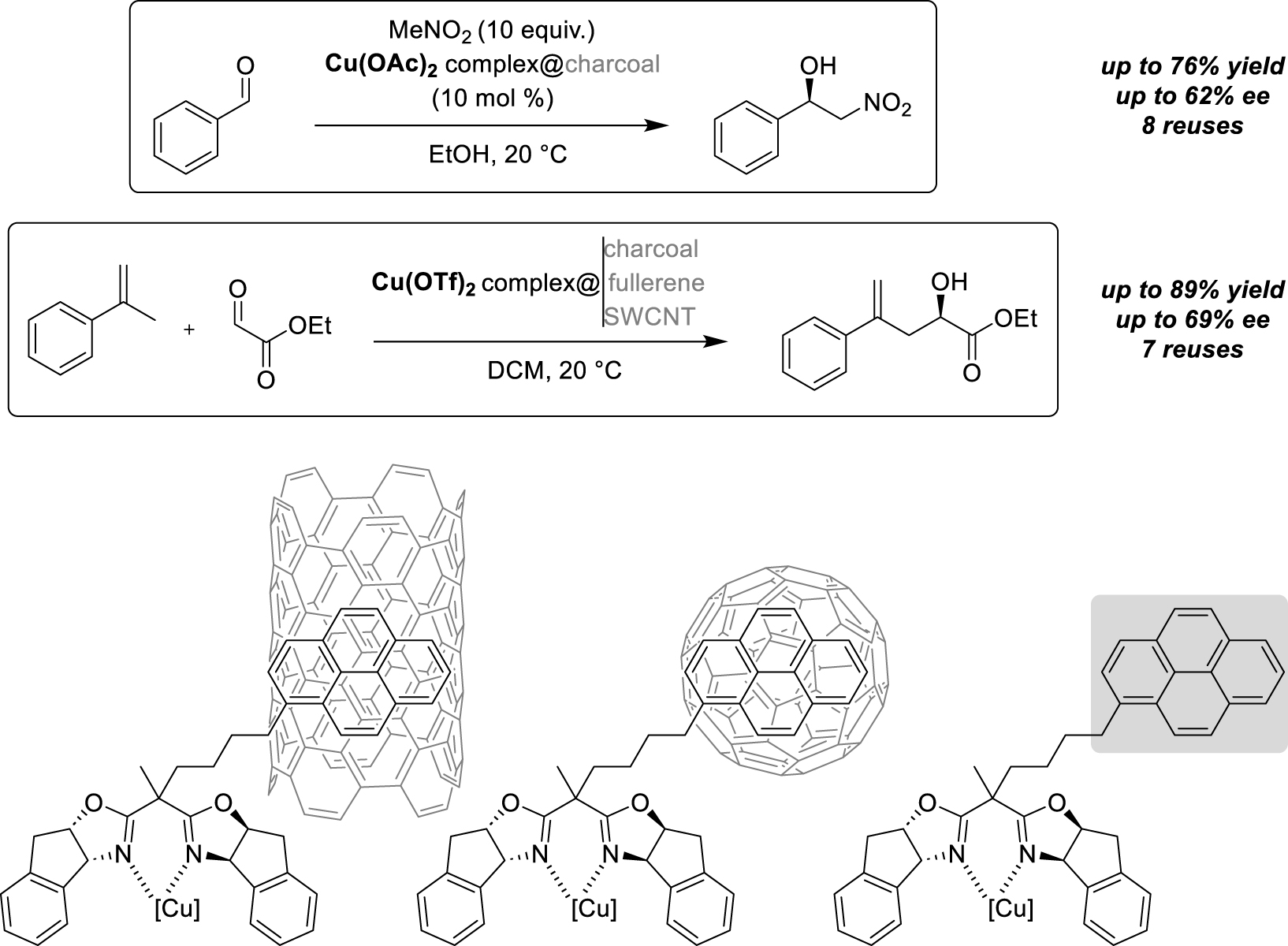
Later, in 2021, Schulz et al. described the immobilization of another type of organometallic complex via a pyrene-tagged salen ligand for both the Asymmetric Ring-Opening (ARO) of a meso-epoxide and the hetero-Diels–Alder reaction (HDA) (Scheme 10) [31]. Herein, different types of chromium salen complexes have been designed and their activity studied for the cyclohexene oxide ARO. Indeed, one- and four-carbon linkers have been synthesized and then, both respective symmetric and unsymmetric ligands have been accessed to finally compare the four ligand-tagged activity. First, in homogeneous conditions, those new-designed catalysts have shown a better activity with improved conversion rates of starting materials compared to the classical Jacobsen ligand, probably explained by possible π-stacking interactions between two catalysts to form bi-metallic more active complexes. However, symmetric catalysts seemed to be considerably less enantioselective than unsymmetric ones. Then, the immobilization of unsymmetric complexes on rGO as a suspension with a 4:1 mass ratio, characterised by UV–Vis and XPS (X-Ray Photoelectron Spectroscopy) analyses, has been performed to switch to heterogeneous conditions. Interestingly, in this case, the catalyst activity was not affected by the heterogenization although the enantioselectivity slightly decreased after the first run. Nevertheless, as the recycling progressed, the enantiomeric excesses increased to reach the same values as the ones obtained under homogeneous conditions. Next, HDA of different aldehydes with Danishefsky’s diene has been then performed to extend the application scope of this new-designed unsymmetrical salen chromium complex immobilized on rGO. Again, the catalyst efficiency was not affected in terms of yields and enantioselectivity with benzaldehyde compared to the use of the classic salen complex. Concerning the immobilization procedure, the nature of the washing solvent which allowed the supported catalyst recovery after one run proved to be very important. Indeed, both cold dichloromethane and acetone afforded a non-negligible leaching of the catalyst which affected the conversion of the starting materials. Diethyl ether finally allowed to perform seven cycles without any impact on either the conversion or the enantioselectivity. Moreover, the authors studied the ability of using the same immobilized-catalyst batch for the transformation of three different aldehydes, successively. Interestingly, the reactivity, with the three different substrates, remained stable even after two runs, showing the robustness of the recovery procedure.
π-stacking interactions on rGO for epoxides aminolysis and hetero Diels–Alder reaction.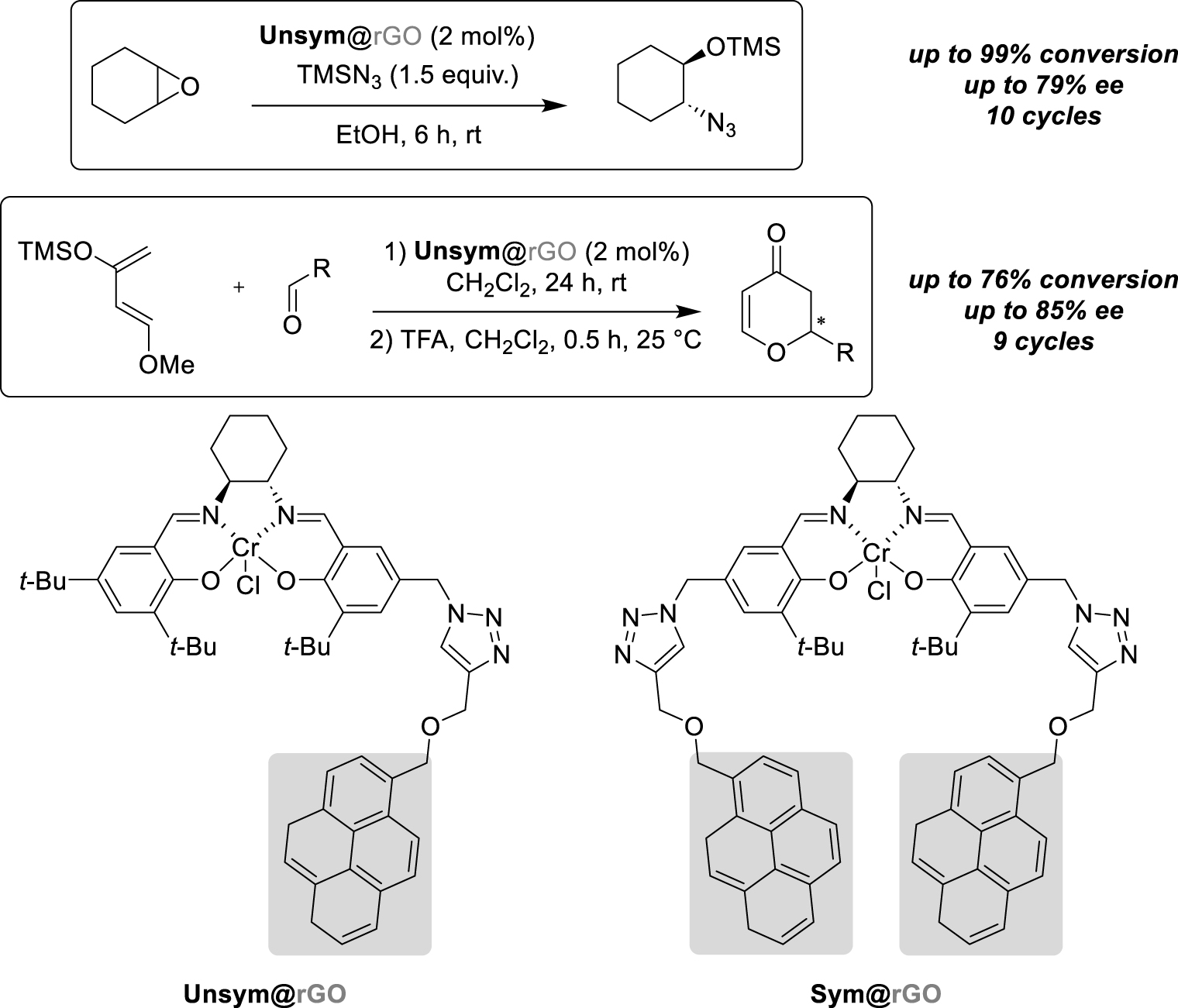
In the same year, Schulz and her group developed heterogeneous conditions to perform the enantioselective formal [3 + 2]-cycloaddition of oxaziridine with ammonium enolates described by Smith group [32]. This reaction was promoted by an IsoThioUrea (ITU) catalyst on phenyl acetic anhydrides allowing to study the heterogenization of the organocatalytic transformation by π-stacking interactions (Scheme 11). Indeed, the organic catalyst has been tagged with a pyrene group on the benzotetramisole phenyl ring and the activity of this newly designed species has been first evaluated under homogeneous conditions. Fortunately, the same high activity as described with the unmodified ITU was recorded and it was shown that using the soluble Hünig base instead of inorganic Cs2CO3 resulted in improving the yields (92%) despite a slight decrease in selectivity (55/45% dr(anti∕syn), 89/89% ee(anti∕syn)) and the formation of a small amount of N-tosylbenzylimine by-product. With those results in hands, the immobilised catalyst on rGO has been engaged and both organic and inorganic bases have been tested allowing to maintain quite the same activity, a stable good activity along three runs (60% yield, dr(anti∕syn) about 50:50, and ee values for both products varying between 85 and 93%). Using dichloromethane was preferred as both reaction and washing solvent. Finally, an extended scope was developed, using four different substituted homoanhydrides, and the same batch of reused catalyst enabled the four different product mixtures to be obtained successively in good yields, with moderate diastereoselectivity and high enantioselectivity values.
π-stacking interactions on rGO for the enantioselective synthesis of oxaziridine derivatives.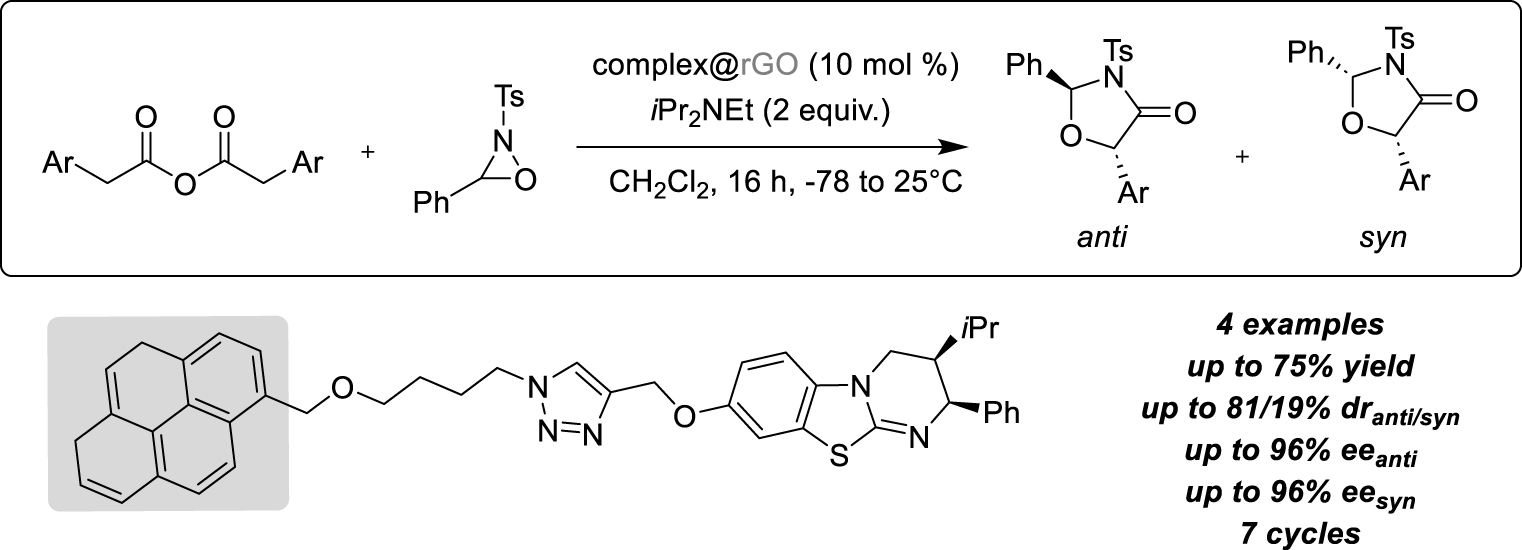
4. Formation of charge-transfer complexes
Schulz and collaborators, using Atomic Force Microscopy (AFM), were able to visualize the formation of weak bonds between two acceptor–donor aromatic molecules, forming Charge-Transfer Complexes (CTC). They indeed successfully functionalized a tip and a probe with electron-deficient TriNitroFluorenone (TNF, (acceptor)) and electron-rich 1-methanolanthracene siloxane (donor), respectively, to quantify the reversible pull-off force between the two species [33]. Furthermore, they showed that CTC interactions could be influenced by the presence of competitive species. For instance, employing an apolar dodecane solvent revealed stronger interactions (6.6 ± 3.5 nN) than using aromatic apolar 1-methylnaphthalene (1.7 ± 0.5 nN), which engaged in competitive π-stacking interactions and saturated the tip-accessible sites, leading to decreased measurements. Various factors, such as time of contact between the species, medium concentration, viscosity, sweep time, and tip geometry, played crucial roles in measuring bonding forces [34]. Building on these results, Schulz et al. exploited CTC interactions for asymmetric catalysis. They developed an innovative recycling method by creating solid CTC between a donor-tagged catalyst and a good acceptor additive, facilitating simple recovery through filtration at the end of the reaction. The first described example involved the design of new anthracene-tagged bis(oxazoline) ligands complexed with Cu(II) to proceed enantioselective Diels–Alder reaction [35]. First, the activity of the new designed complex was evaluated under homogeneous conditions in dichloromethane and showed no impact on both conversion (>99%) and selectivity (93% de, 84% ee) after the first run, for the reaction between cyclopentadiene and 3-acryloyl-oxazolidin-2-one. CTC formation was then achieved by adding TNF, and the resulting complex precipitated thanks to pentane addition, enabling easy catalyst recovery for subsequent runs without compromising activity. Switching to another dienophile with the same recovered catalyst allowed for five more cycles at 20 °C. Eleven runs were successfully processed with the same batch, recovered by precipitation, and washing with pentane, without impacting yields and enantioselectivity (Scheme 12A). The following year, in 2007, the method was extended to three newly designed bis(oxazoline) ligands in the Cu(II) promoted Diels–Alder reaction [36], involving one (R)-valinol and two tert-leucinol bis(oxazoline) derivatives, each tagged with anthracene. At first, the reaction was conducted under homogeneous conditions to assess the impact of ligand modification on catalyst activity. Concerning the (R)-valinol derivative, there was a significant decrease on selectivity (22% ee), mainly explained by the partial loss of the C2 symmetry of the ligand after functionalization on the gem-dimethyl group compared to classical bis(oxazoline). Then, regarding the catalyst recovery, CTC were prior prepared ex-situ before being introduced to the reaction solvent with substrates. Fortunately, over six runs, the recovered catalyst was re-engaged with new substrates without compromising the enantioselectivity. In parallel, similar study was conducted with the two tert-leucinol derivatives as ligand (three- and five-carbon linker) which appeared to have a lesser impact on enantioselectivity than the first one (97% ee after one run under homogeneous conditions). However, initial tests in the presence of the corresponding CTC-based catalysts were much less effective (15 and 33% ee). The reuse for a second attempt with the same CTCs and new substrates surprisingly resulted in significantly improved enantiomeric excesses in both cases. Notably, the addition of molecular sieves was mandatory to prevent the detrimental effect of water on copper complex. PM3 calculations also revealed that in these latter complexes, the distortion degree between tert-leucinol-derivative ligands and the catalytic site was larger than with (R)-valinol one, involving more steric hindrance around the copper center. To overcome this activity loss, it was demonstrated that the first run needed to proceed without the electron-deficient precursor to achieve better results (89% yield, 87% ee), and the formation of the CTC must be executed only at the end of the first transformation. By adopting this approach, improved yields (89%) and enantiomeric excesses (87% ee) were achieved after the second run with the same catalyst batch and remained stable along two additional cycles. Finally, three more cycles were conducted by switching from previously used 3-(but-2-enoyl)-oxazolidin-2-one to 3-acryloyloxazolidin-2-one, sustaining satisfactory activity (Scheme 12B). In 2008, our group proposed to use CTC interactions to prepare mimic bidentate complexes from anthracene- and TNF-tagged monodentate bis(oxazoline) [37]. We focused on the design of reversible copper(II) catalysts from copper triflate for use in asymmetric Diels–Alder cycloaddition. Initially, unsymmetrical modified bis(oxazoline) was prepared by deriving from (S)-valinol, (R)-phenylglycinol and (S)-tert-leucinol. These three ligands were respectively tagged with both anthracene as a donor and TNF as an acceptor. With these six ligands in hand, catalytic tests were attempted under homogeneous conditions to evaluate the tag impact on reactivity. Fortunately, good conversion and enantioselectivities were achieved using monodentates species independently, resulting in major formation of the endo-product with the anthracene-tagged electron-rich ligand and exo-product with the TNF-tagged electron-deficient one. This phenomenon was particularly evident with tert-leucinol derivatives and was explained by likely π-stacking interactions between the tags, favoring the formation of homodimeric copper complexes well known to efficiently catalyze asymmetric transformations. Subsequently, mixtures of one electron-rich and one electron-deficient ligand equivalent (1:1) afforded heterodimeric CTC. Unfortunately, as previously demonstrated [33, 34], competitive π-stacking could occur between aromatic moieties, leading to the formation of homo-and hetero-dimeric bidentate complexes, resulting in poor yields and enantiomeric excesses for the endo and exo products. Nevertheless, a method has been developed for easily recovering the catalyst by precipitating the CTCs, facilitated by a simple pentane addition. The reuse of those CTC complexes did not affect conversion and enantioselectivity in two runs, demonstrating the efficiency of the procedure for recycling (Scheme 12C).
Immobilization by CTC interactions for the Diels–Alder reaction.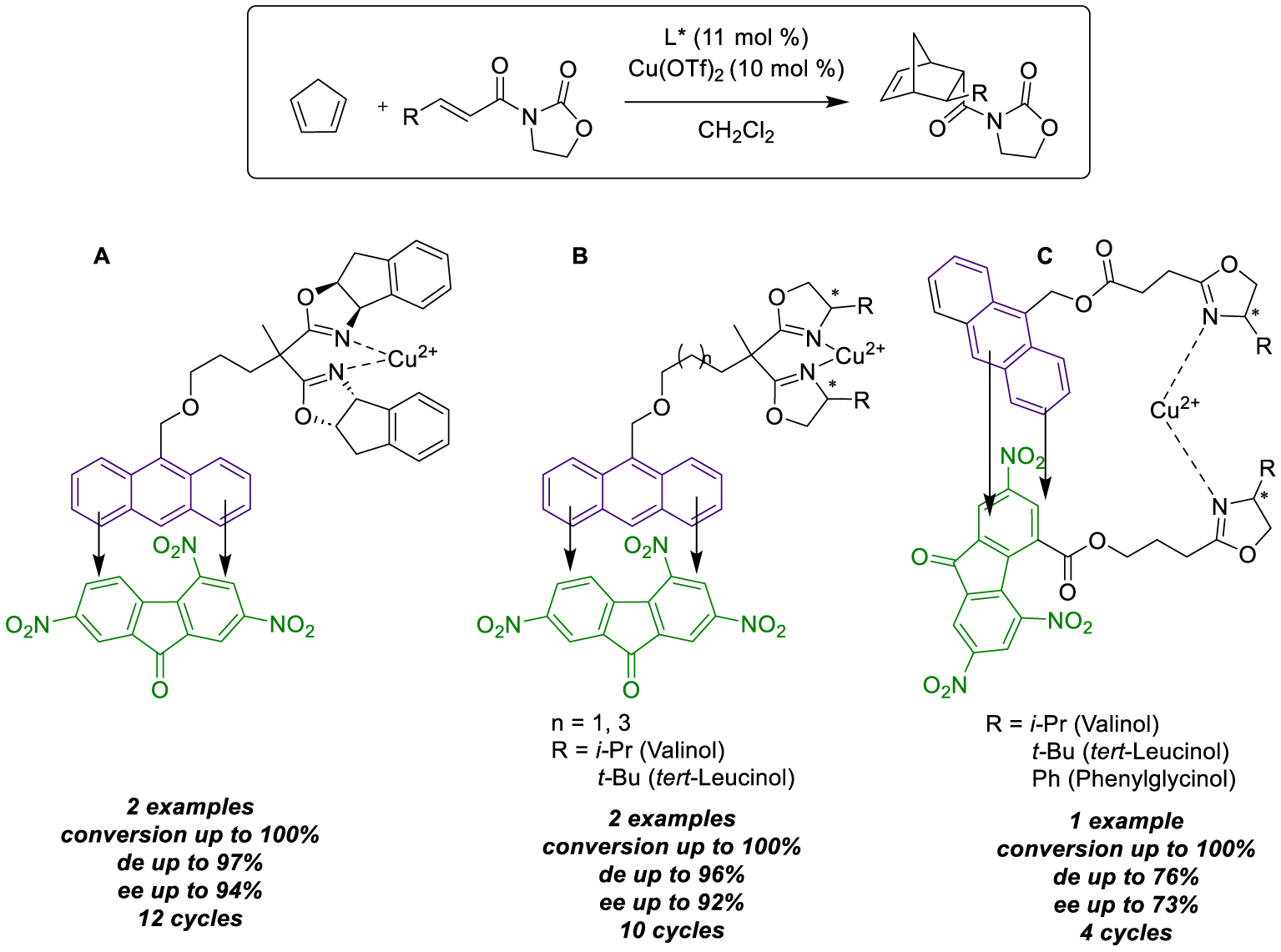
In the same year, Gennari et al. also reported the formation of bidentate complexes via CTC interactions between modified ligands, applied to Rh-catalysed hydrogenation [38]. They first prepared a library of tagged-ligands, consisting in eight electron-rich (six BINOL-and two octahydro-BINOL-based) and two electron-deficient pentafluorobenzyl phosphites (R and S). After isolating these compounds, they engaged them separately into a solution with the Rh(cod)BF4 pre-catalyst (L/Rh = 2:1)to form homodimeric complexes, characterized by 31P NMR. In parallel, they applied the same method to obtain heterodimeric CTC by mixing an equimolar ratio of electron-rich, electron-poor, and Rh pre-catalyst. In these cases, they observed the formation of the desired hetero-CTC (50%), but unfortunately, also the formation of homo-complexes (50%) due to competitive interactions. Consequently, they decided to conduct attempts with homodimeric and heterodimeric complexes in the enantioselective hydrogenation of dimethyl itaconate (Scheme 13). The best outcome was achieved with the mixture of (R)-electron-poor phosphite and (R)-meta-dimethoxybenzylalcohol-modified electron-rich ligand (99% ee). Other heterodimeric derivatives yielded moderate to excellent enantioselectivities, generally exhibiting higher activity with (R)/(R) mixtures compared to (R)/(S) ones. Additionally, concerning homodimeric species, methoxyphenol derivatives led to improved enantioselectivities (92–99% ee) compared to methoxybenzylalcohol ones, which delivered poor enantiomeric excesses (70–84% ee) and octahydro-BINOL ones, which provided moderate values (50–54% ee). Finally, they applied their library of bidentate complexes to the same reaction on methyl acetamido acrylate and, again, heterodimeric (R)-dimethoxybenzylalcohol derivative/(R)-pentafluorobenzyl one was the most successful trial (93% ee). Despite the heterodimeric catalyst not being pure, the corresponding heterodimeric/homodimeric mixture turned out to be the most active catalytic species.
CTC interactions for hydrogenation reactions.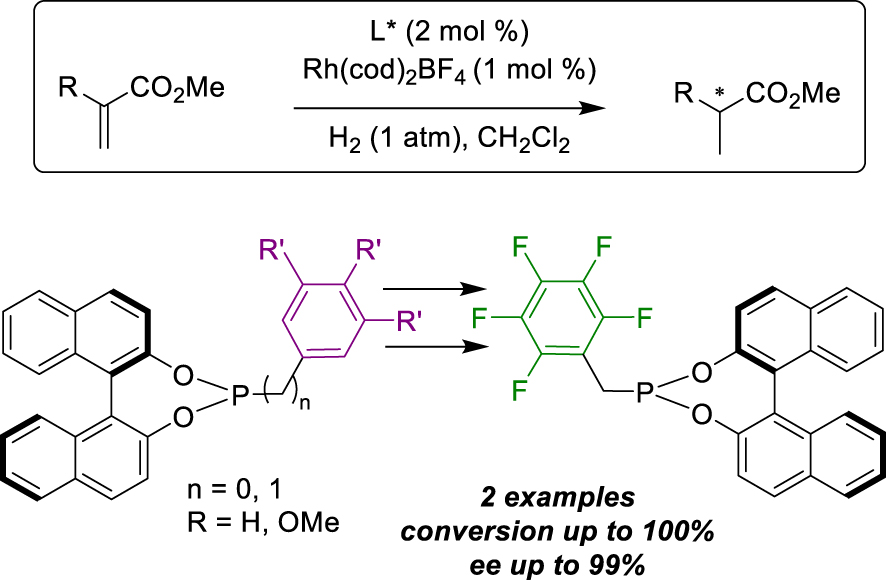
In 2010, the Schulz group described a new immobilization method based on the same CTC interactions but involving the use of a modified-resin solid support [39]. In this case, they thought to functionalize a Merrifield resin with a TNF moiety as an acceptor for immobilizing anthracene-tagged ligands. The advantage of this method is that it facilitates catalyst recovery and allows one catalyst to be replaced by another to carry out different reactions in sequence, while using the same support. The reaction chosen to highlight this strategy was the Cu(II) enantioselective Diels–Alder reaction between 3-but-2-enoyl-oxazolidin-2-one and cyclopentadiene. Initially, the efficiency of the newly designed tagged ligand was evaluated under homogeneous conditions without any detrimental effect on activity (83% ee, 78% de). Subsequently, the newly formed Cu(II)-catalyst was engaged under the same reaction conditions after its immobilization was conducted ex situ by CTC formation with the modified resin (0.5 mmol/g TNF loading, <50 mesh particle size). Once again, the same activity was achieved, leading to the desired endo product as the major compound (91% yield, 76% ee). Five additional runs were achieved using the same immobilized catalyst batch, recovered by simple filtration, and washing with pentane. During the first two reuses, the activity seemed stable, but from the fourth recycling, the yield and selectivity began to decrease, reaching very low values after the sixth reuse (28% yield, 39% ee). Test reactions indicated a partial leaching of the catalyst and weak CTC interactions (Scheme 14A). Finally, the same support was tested for different reactions; to this end, the first attempts were run with a single anthracene-tagged catalyst over four successive runs, revealing a loss of activity during the last two runs. To change the catalyst, toluene was introduced as a competitive solvent for CTC interactions, facilitating the removal of the catalyst from the solid support. The ent-catalyst was then easily added in dichloromethane to bind it to the recovered TNF-tagged solid support, and the same antagonistic reaction was conducted. Four additional runs were carried out using the same conditions, furnishing the opposite enantiomer of the target product with the same enantiomeric excesses as before. This method has proved to be an effective approach for the recycle of the catalysts and conducting multiple reactions using the same support, which could prove particularly useful in flow chemistry, for example. The same group also described another multi-reaction approach facilitated by CTC assistance for Cu(II)-catalyst recycling [40]. In this context, the aim was to use the same catalyst batch for the asymmetric Henry nitroaldolization, applied to multiple substrates. The study focused on an anthracene-tagged bis(oxazoline) ligand, as previously described, complexed with Cu(OAc)2⋅H2O. The presence of the anthracene moiety was proven to be not detrimental to the reaction activity (89% yield, 90% ee). Conveniently, catalyst recovery was achieved by adding TNF for CTC formation and subsequent precipitation in pentane at the end of the first run. After one recycling, the enantioselectivity remained stable, indicating that the CTC formation had no negative impact on the catalyst activity. Notably, the reaction time increased over the other runs, probably due to a leaching of the catalyst, but six reuses were successfully conducted while maintaining good results. This method was then applied to various substrates, including five aldehydes and two nitroalkane derivatives, resulting in seven different products with good yields and enantiomeric excesses. Especially, reactions involving nitroethane yielded a diastereoisomer mixture, with the anti-isomer as the major product. With these results in hands, they accomplished the recycling of one catalyst batch for nine different transformations, demonstrating that no impact on selectivity was observed over the runs. Despite a slight increase in reaction time during the subsequent runs, it is important to note that no traces of the previous product were observed during the next run, indicating that the recycling and washing procedures had been well optimized. Finally, they switched to heterogeneous conditions by grafting TNF moieties onto activated silica and immobilizing an anthracene-tagged Cu(II) catalyst via CTC interactions. The Henry reaction could be performed five successive times with the same catalyst batch, without additional precipitation in pentane, affording the desired product with the same yields and enantioselectivity values as under homogeneous conditions (Scheme 14B). This method represents another original example of immobilization and recovery of catalysts on a solid support by non-covalent CTC interactions.
Immobilization by formation of CTC with organic and inorganic supports.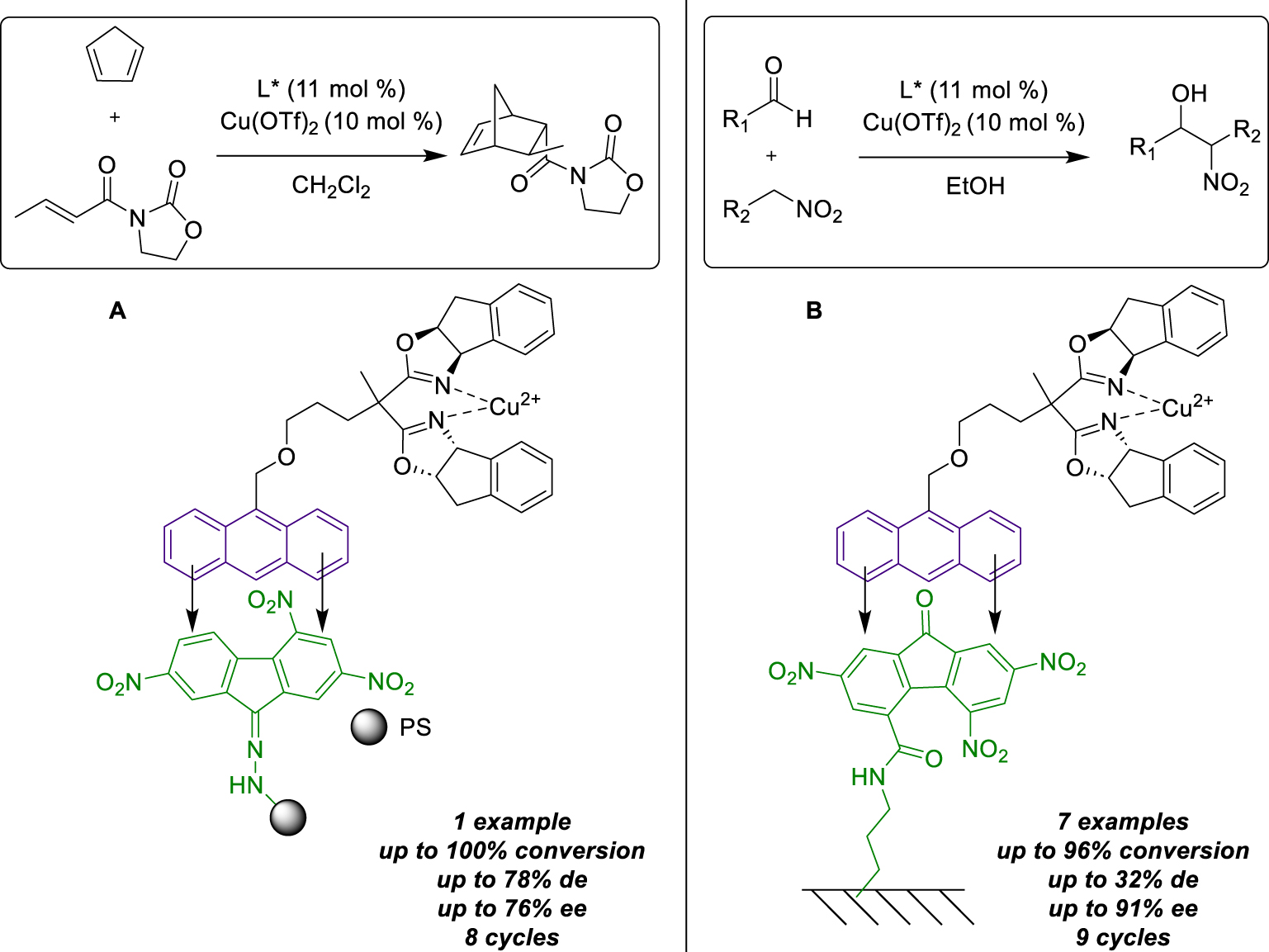
In 2011, our group proposed a new asymmetric multi-reaction method facilitating the promotion of additional Cu-catalyzed transformations, namely ene and cyclopropanation reactions [41]. These transformations were already known to process by bis(oxazoline) ligands with Cu(OTf)2. This study reported the use of anthracene-modified bis(oxazoline) for the easy recovery and recycling of the catalyst. The ene reaction was firstly carried out under homogeneous conditions between α-methylstyrene and ethyl glyoxylate, resulting in a similar yield (89%) and enantiomeric excess (67% ee) as with the parent symmetrical ligands. Secondly, after the first run and TNF addition, a further nine cycles of catalyst recycling were carried out with no effect on catalyst activity. CTC Precipitation in pentane was employed to ensure effective recovery and similar results were achieved on the analogous 3,3,3-trifluoro-2-oxo-propionic acid methylester over seven reuses using the same method. Likewise, the cyclopropanation of styrene with ethyl diazoacetate was investigated with the same Cu-complex. The initial trial also showed a good yield (69%) with a mixture of diastereoisomers (2:3 ratio) and good enantiomeric excesses (72% and 69% ee for each diastereoisomers), in agreement with the results obtained using conventional homogeneous catalysts. These results were replicated over five cycles, with a slight loss in activity observed from the sixth run (41% yield, 67 and 69% ee). The main objective of this work was therefore to execute these different reactions using a single catalyst batch, thanks to the precipitation of CTC for catalyst recycling (Scheme 15). As previously reported [35, 36], Diels–Alder cycloaddition was also achieved via this procedure and was consequently considered. The reaction was first carried out twice between 3-acryloyloxazolidin-2-one and cyclopentadiene, then twice more times between 3-(but-2-enoyl)-oxazolidin-2-one and the same diene to yield another derivative. Subsequently, the ene reaction was conducted three times with α-methylstyrene and ethyl glyoxylate, yielding good results. Finally, the same batch of catalyst was again used for cyclopropanation, giving results in line with those described above. This unprecedented multi-reaction approach enabled the same catalyst batch to be reused nine times without any loss of activity in successive reactions with different substrates.
CTC interactions for an asymmetric multi-reaction procedure.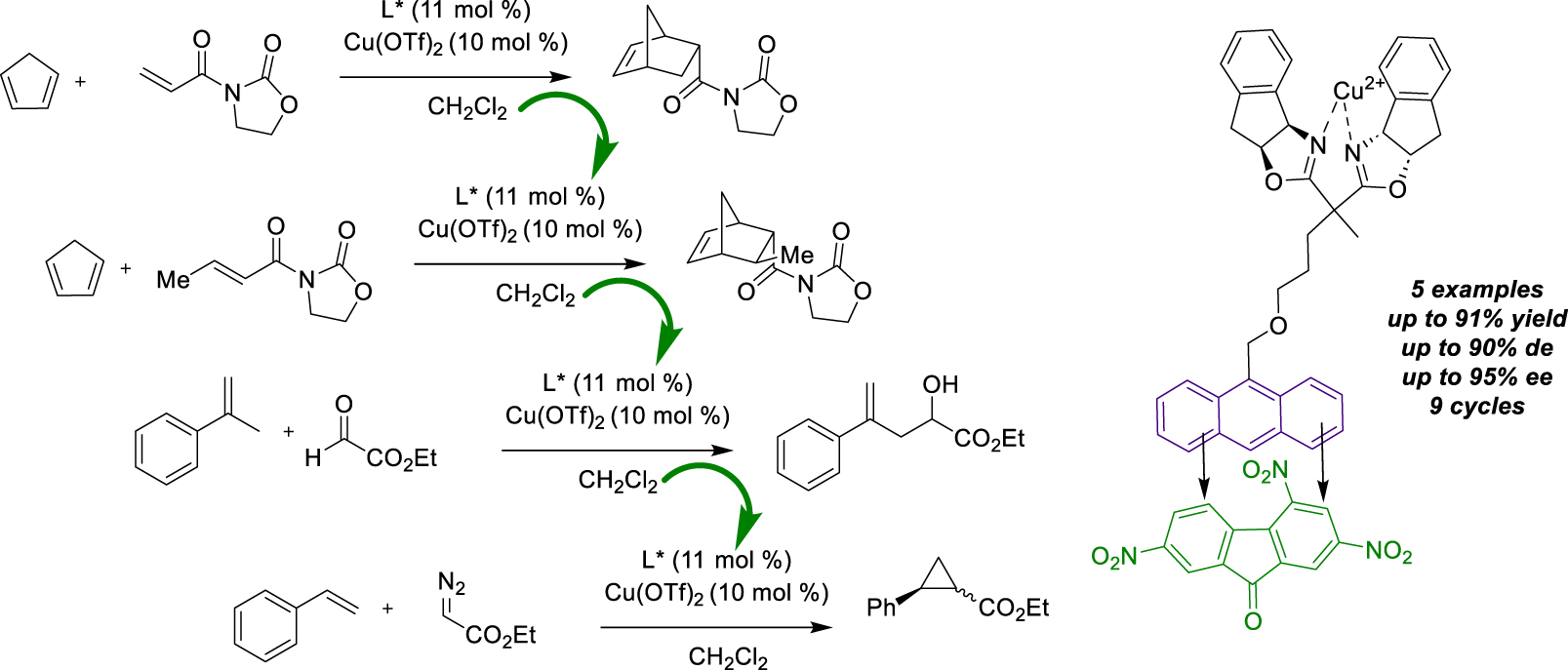
On another hand, in 2012, Schulz and Didier described the immobilization of the same bis(oxazoline) structures but tagged with an electron-deficient TNF moiety [42]. Immobilization via CTC interactions was therefore considered by adding anthracene after the first reaction and introducing apolar pentane to precipitate the formed CTC, facilitating a good catalyst recovery. The first attempt involved the TNF-modified ligand and Cu(OTf)2 in the well-known Diels–Alder cycloaddition between cyclopentadiene and 3-but-2-enoyloxazolidin-2-one. At the end of the reaction, a precipitation procedure and filtration were conducted to recover the solid. Surprisingly, all the anthracene was recovered in solution, indicating that the interactions with the CTC were not sufficiently strong. In fact, regarding the solubility of the TNF-tagged Cu-complex in different solvents, it precipitated on its own when the solvent was changed, and the addition of anthracene for the CTC formation was not necessary for the catalyst recovery. Subsequently, the solid TNF-complex was reused with new substrates and proved effective in fourteen runs with the same reactant, followed by a further six runs with analogous 3-acryloyloxazolidin-2-one. A lower temperature was required with this latter substrate to inhibit competitive racemic transformation. More recently, in 2019, Liang and collaborators described the use of CTC in the Hydrolytic Kinetic Resolution (HKR) of epichlorohydrin, catalyzed by Co(III)-salen complexes [43]. Since this transformation involves a bimetallic system, where one metal acts as a Lewis acid to activate the epoxide and the other to activate the hydroxide, the formation of donor–acceptor complexes to bring two salen molecules together was found to be highly beneficial. Modification of the symmetrical salen-ligands with two electron-poor diimide tags facilitated interactions with additional polyaromatic species, allowing the formation of acceptor–donor–acceptor CTC sandwiches at an ideal distance of around 7–8 Å, optimal for enhancing HKR reactions. The group synthesized three generations of modified salen ligands to explore various parameters. Their results revealed that flexible alkyl ester linkers between the acceptor and the salen core were more efficient than rigid conjugated phenyl linkers, enabling favorable orientation and geometrical arrangement of the bimetallic system. In addition, the choice of polyaromatic donor species played a crucial role, improving the catalyst activity when used in the proper quantity. In this case, the best results were obtained with the symmetrical di-Pyromellitic DiImide (PDI) acceptor in the presence of pyrene or perylene (conversion rates of 52% and 54%, >99% ee, respectively), outperforming the classical Jacobsen salen (10 h vs. 42 h). The face-to-face stacking of the bimetallic CTC probably optimally aligned the Co(III)-salen cores, optimizing the interactions between both substrates. This was supported by 1H NMR and UV–Vis studies, highlighting donor–acceptor interactions between PDI and pyrene moieties (Scheme 16A). In addition to this, the same group reported another CTC-immobilized bimetallic system for the same reaction, expanding the scope to other epoxides and nucleophiles [44]. They designed new modified salen ligands, including unsymmetrical Naphtalene DiImide (NDI)/pyrene-bi-tagged, mono-NDI or mono-pyrene, and symmetrical bi-NDI or bi-pyrene-tagged ligands, comparing their activity with the non-substituted Jacobsen complex. In the epichlorohydrin HKR, all catalysts led to improved yields (50%–59%) and enantioselectivity (>99%) compared to the classical salen, with particularly decreased reaction rates obtained in the presence of the unsymmetrical acceptor–donor tagged ligand (3.5 h). Another noteworthy attempt, involving a mixture of both mono-substituted acceptor and donor (1:1 ratio), yielded similar results (59% yield, >99% ee). Further experiments involved committing both CTCs to other terminal epoxides HKR, which gave satisfactory results, with slightly better activity observed with the mixture of two differently tagged complexes, particularly on aliphatic and hindered substrates. It also allowed to reduce reaction time for the transformation of 1,2-epoxybutane and 1,2-epoxyhexane epoxide without affecting the selectivity. The higher activity was attributed to a more flexible bi-complex between two mono-tagged than two bi-tagged ones, allowing an optimized arrangement in the space between the two salen catalytic moieties (Scheme 16B). Overall, these results were also observed on more complex substrates, demonstrating good solvent adaptability, high catalytic activity, and enantioselectivity in the asymmetric ring opening of epoxides.
CTC formation for ARO of epoxides.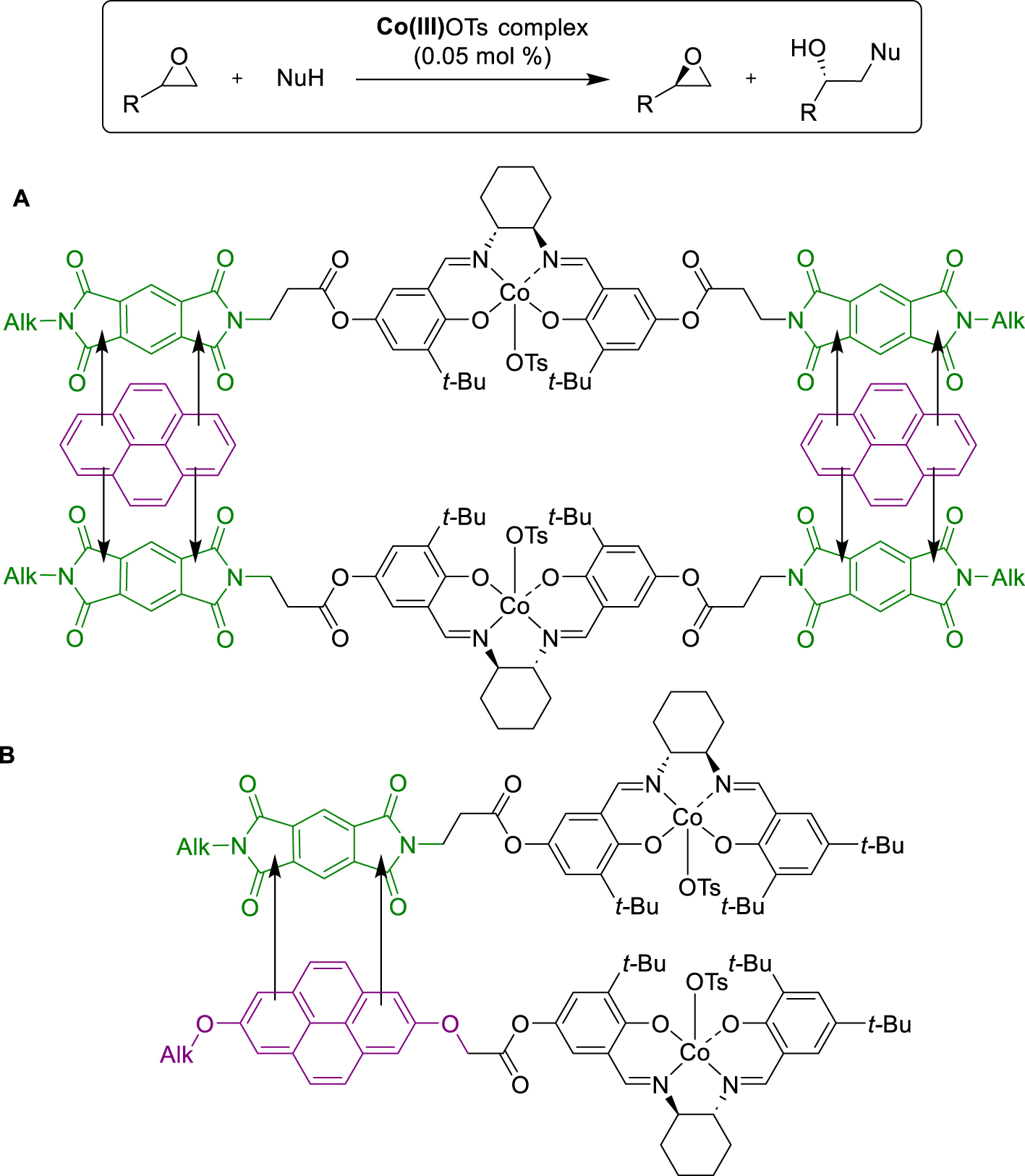
5. Coordination links—Metal Organic Frameworks (MOF)
Another strategy has been developed for the immobilization of chiral organometallic catalysts involving the heterocombination or homocombination of different mutitopic ligands with metal ions, leading to self-supported catalysts, active as heterogeneous species, or to self-supported dynamic catalysts, mainly effective as homogeneous species [45]. To illustrate the first case, Ding and coworkers prepared two different ligands both possessing ditopic sites in the form of a bridged BINAP and a bridged diphenyldiamine (Scheme 17) [46]. After coordination of ruthenium in the presence of the two ligands, a one-dimensional structure is formed as a self-supported Noyori-type catalyst, in the form of an iso-propanol-insoluble light-brown solid. The selectivity of this catalyst proved superior to that of its homogeneous counterpart, promoting the hydrogenation of acetophenone and seven other arylketones with enantioselectivities in excess of 94.5% ee, thus demonstrating the synergistic impact of the supramolecular self-supported assembly on the catalytic behavior. The insoluble catalyst was recovered by filtration and reused seven consecutive times without losing its efficiency.
Immobilization by heterocombination of mutitopic ligands for asymmetric hydrogenation.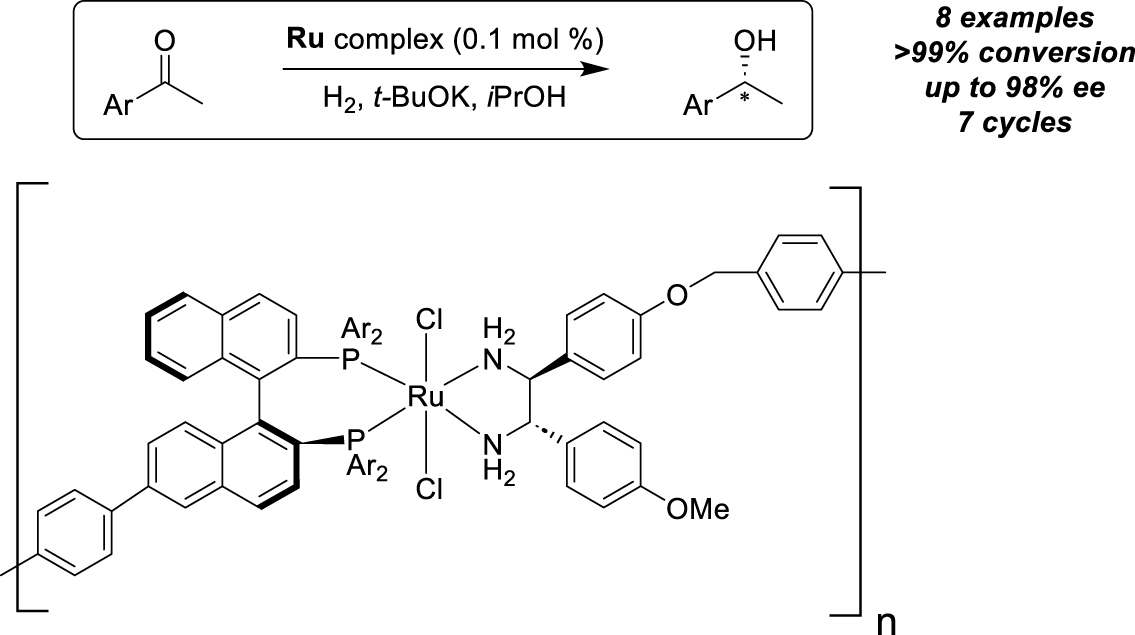
Another related approach involves the homo-assembly of multitopic ligands by metal coordination leading to a polymeric structure. What is looked for in this case, is a catalyst that remains heterogeneous in its resting state at the end of catalysis, yet capable of undergoing a reversible partial de-coordination process in the reaction mixture to become the active species. This concept was demonstrated by García et al., who prepared a new chiral ditopic ligand bearing two azabis(oxazoline) groups (Scheme 18) [47]. Using copper salts of weakly coordinating anions with an equimolar amount of the ditopic ligand in a non-coordinating solvent, formation of the desired coordination polymer occurred. The reaction tested involved the cyclopropanation of styrene with ethyldiazoacetate, as a strongly coordinating species, leading to a de-coordination process upon its addition and a reaction taking place in the homogeneous phase with even higher enantioselectivity values for both cyclopropane isomers (ee up to 97% for the trans product). After completion of the reaction, the coordination polymer is formed again, and can be easily recovered by filtration for its efficient recycling (14 cycles).
Self-supported dynamic copper catalysts for cyclopropanation reactions.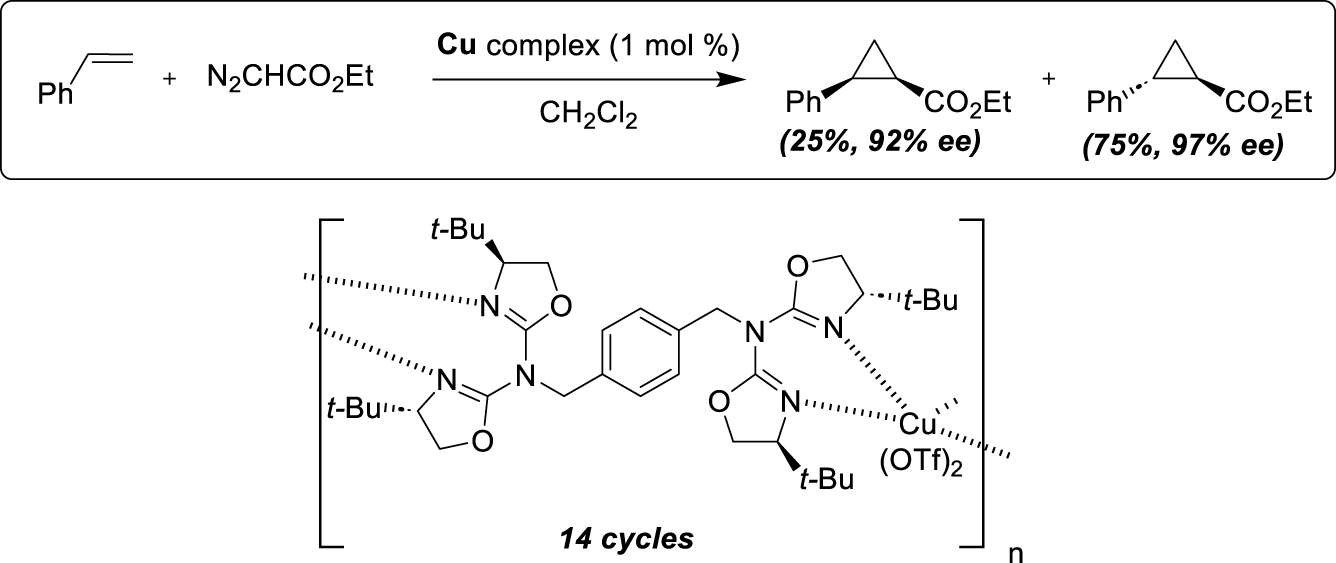
This procedure was subsequently studied by the Bellemin-Laponnaz group for the enantioselective α-hydrazination of β-ketoesters with polytopic (bis-, tris- and tetratopic) bisoxazoline as ligands [48]. The use of the ditopic ligand in combination with Cu(OTf)2 led to a highly efficient self-supporting catalyst for reacting with acyclic and cyclic β-ketoesters (5 examples) delivering the desired products with almost complete conversion and enantioselectivity values of 99%. Depending on the substrate structure, six to ten runs could be performed without loss of efficiency. Interestingly, increasing the topicity of the ligand led to a worse recycling and more catalyst leaching. The authors also studied NonLinear Effects (NLE) to better understand the aggregation state of the active species, conducting experiments in which the optical purity of the ligand varied. Negative NLE was observed, consistent with the preferential formation of a homochiral metallopolymer, in equilibrium with the active monomeric species. These authors applied further this concept for the preparation of air- and moisture-stable nickel metallopolymers efficient as enantioselective catalysts for the Michael addition of diester malonates to nitroalkenes [49]. A chiral ditopic cyclohexyldiamine-based ligand was prepared generating a self-supported catalyst in the presence of nickel salts (Scheme 19). The substrate displaces a diamine ligand and generates an active catalyst that contains only a single diamine ligand, giving an efficient catalyst with greater activity than the corresponding monomer and similar enantioselectivity. Five examples were described (complete conversion and up to 93% ee), along with a procedure for efficient recovery of the catalytic species. It involves the addition of diethylether for catalyst precipitation, filtration, washing and subsequent reuse in up to 13 cycles with stable enantioselectivity and a slight decrease in substrate conversion.
Ditopic cyclohexyldiamine-based ligand as self-supported nickel catalyst.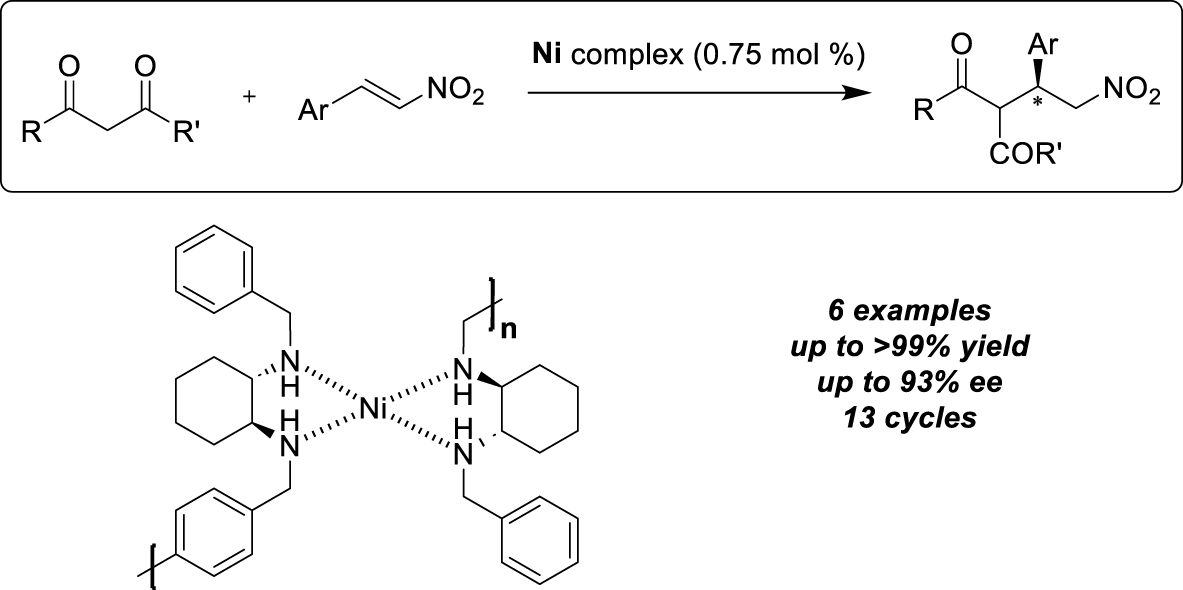
For over fifteen years now, metal organic frameworks (MOFs) have also been studied as a response to certain challenges in supported asymmetric catalysis [50, 51]. The activity of these heterogeneous catalysts comes either from unsaturated metal coordination sites, or from active linkers between metals, or both, particularly if these linkers are chiral organometallic complexes. In addition, the wide choice of porous structures available allows selective limitations in terms of size and shape through well-defined channels and pores. Thus, MOFs have been reported fairly recently as effective heterogeneous catalysts for a number of transformations, including enantioselective reactions. A relatively early example was described by the Lin’s group for the preparation of homochiral porous MOFs from BINOL derivatives as chiral bridging ligands containing bipyridyl units as orthogonal functional groups (Scheme 20) [52]. They were able to produce colorless crystals in the presence of CdCl2 while the chiral dihydroxy groups were accessible for reaction with Ti(Oi-Pr)4, enabling the formation of an active heterogeneous catalyst for the addition of Et2Zn to aromatic aldehydes. The authors showed activity and enantioselectivity equivalent to those obtained when homogeneous BINOL was used as a ligand (8 examples compared), except for some substrates bearing Frechet-type dendrons, which were probably unable to access the catalytic sites because they were too large. This result proved that catalysis occurred inside the MOF pores, and control experiments were also carried out to confirm the heterogeneous nature of the reaction.
BINOL-based homochiral porous MOF for the ZnEt2 addition on aldehydes.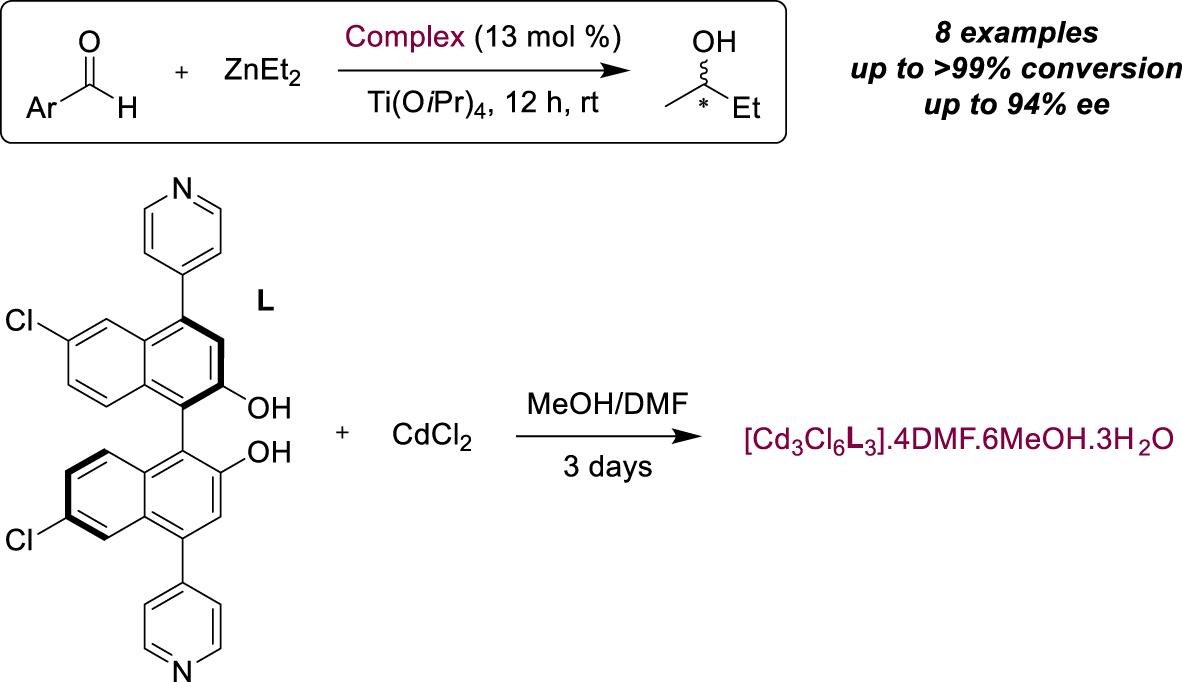
One year later, Nguyen and Hupp prepared another microporous MOF with Mn-salen struts for asymmetric epoxidation [53]. The Mn-salen was modified with two pyridine units, and a robust structure, in the form of a pair of interpenetrating networks, was obtained from zinc salts associated with biphenyldicarboxylate as the second ligand. For the asymmetric epoxidation of chromene derivatives and 2-(tert-butylsulfonyl)iodosylbenzene as oxidant, the heterogeneous catalyst proved to be almost four times more active than the corresponding homogeneous catalyst, with only a slight decrease in enantioselectivity (from 88 to 82% ee). Simple recovery of the MOF catalyst by filtration enabled it to be reused for three cycles without any decrease in enantioselectivity and with a slight loss of activity, probably due to partial fragmentation of the particles.
Since these early examples, a great deal of work has been devoted to the precise optimization of the MOF structure in order to enhance its reactivity and selectivity according to the target. For instance, a family of isoreticular chiral MOFs has been prepared under solvothermal conditions from Mn-salen derivatives bearing dicarboxylic acids functions attached by spacer bonds of different sizes [54]. The corresponding materials therefore had different structures depending on the solvent used to synthesize them and adjustable open channel sizes, which in some cases led to heterogeneous catalysts that were as effective as homogeneous ones for the asymmetric epoxidation of alkenes, while at the same time being recoverable and reusable after simple filtration.
The versatility of these crystalline structures for asymmetric heterogeneous catalysis was demonstrated by Liu and Cui, who prepared a porous Cr-salen MOF (Scheme 21) and subjected it to a series of four catalytic transformations involving different mechanisms [55]. The crystalline catalyst was obtained by heating the corresponding salen complex with CdI2 and NaOAc and was tested in the Nazarov reaction of alkoxydienones, affording functionalized cyclopentenones with high selectivity. Although the enantioselectivity remained slightly lower than the value obtained with the homogeneous analog, a higher diastereoselectivity was observed, which was attributed to the steric constraints of the confined network. Used in other reactions such as the asymmetric aminolysis of trans-stilbene oxide, the Diels–Alder and hetero-Diels–Alder reactions, it has led to increased enantioselectivity values compared with homogeneous cases, with confinement effects allowing ideal location of the active sites. Size selectivity was also demonstrated as a bulky benzaldehyde derivative could not undergo an hetero-Diels–Alder reaction, indicating a unique catalytic activity within the MOF pores. The catalyst was reused in four aminolysis cycles, showing good stability.
Porous Cr-salen MOF efficient for four catalytic transformations.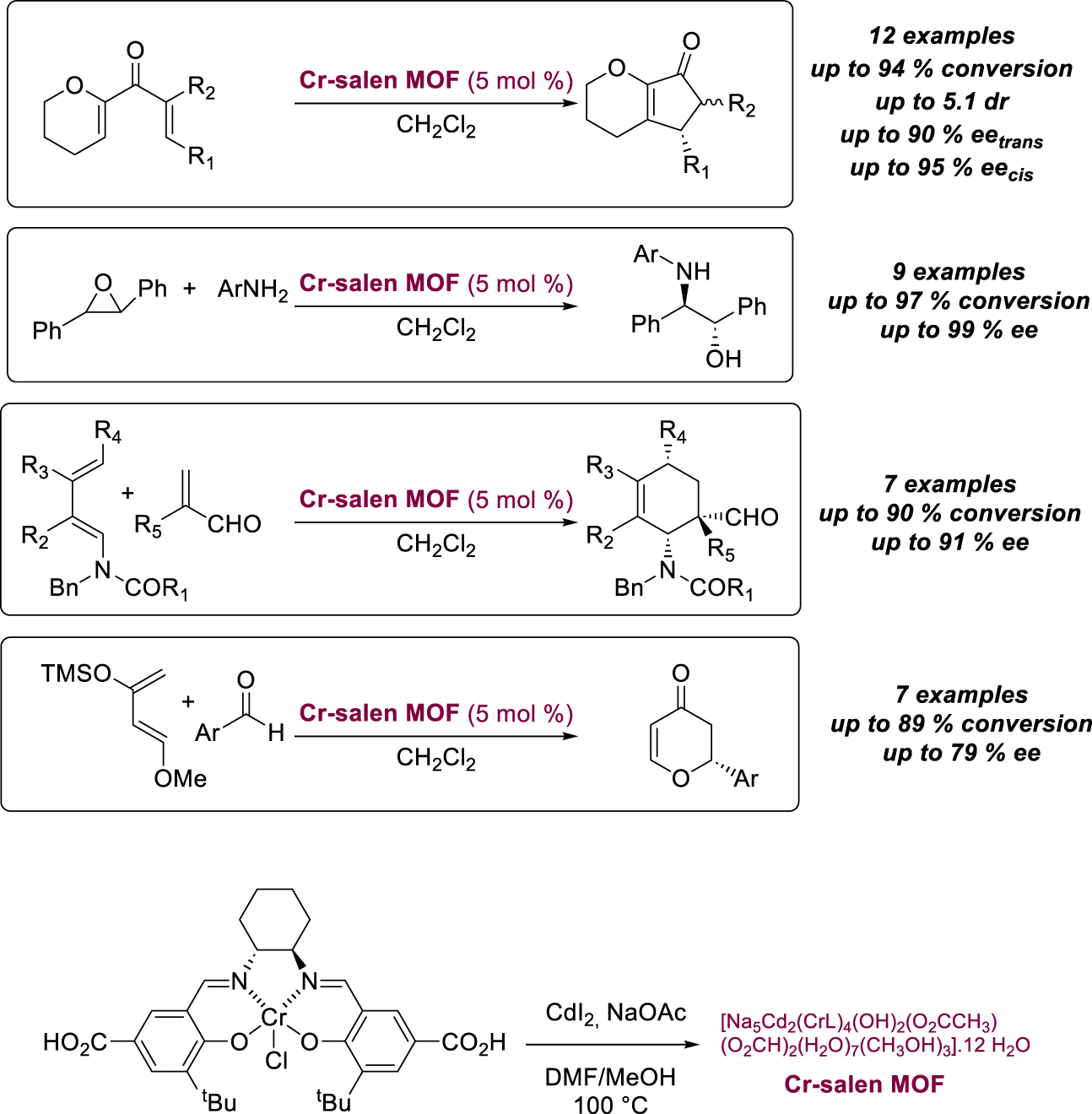
Other advances involving the manipulation of such heterogeneous catalysts concern their post-synthesis modification. Ren and Jiang, for instance, synthesized and fully characterized a 3D Cu-salen MOF possessing a 1D open channel [56]. They then carried out a reduction step in the presence of NaBH4, leading to the reduction of the salen imine groups to a more basic and flexible skeleton, while retaining its structural integrity. Although the Cu-salen MOF proved to be a poor catalyst in the Henry reaction, the corresponding salan derivative afforded the expected products with higher activity and enantioselectivity (15 examples and ee up to 98%) and was successfully recovered and reused for five successive runs. Another post-modification of an existing MOF was described by Liu and Cui, when they heated a VO-salen MOF in the presence of a Cr-salen complex with exactly the same structure; this solvent-assisted linker exchange resulted in the partial replacement of certain VO-salen units by Cr-ones as shown by Inductively Coupled Plasma – Optical Emission Spectrometry (ICP-OES analyses) [57]. While the first catalyst was active in the cyanosilylation of aldehydes (with up to 95% ee, 11 examples), the MOF exchanged by Cr-salen was engaged in asymmetric kinetic resolution with aniline, a reaction that is not catalyzed by VO-salen complexes. Cis-stilbene oxide was efficiently converted (87%) to aminoalcohol (76% ee), demonstrating the introduction, by exchange, of chiral Cr-salen species leading to mixed MOF, promising for cooperative or tandem catalysis. To illustrate cooperative catalysis, the authors demonstrated the presence of a synergistic pathway resulting from the close location of two V-salen units in the open channels of a MOF [58]. They prepared two different heterogeneous MOFs catalysts resulting from Cd(NO3)2 and a VO-salen complex or a mixture of a Cu- and a VO-salen complex. Once the two networks had been fully characterized, they were used in the test reaction involving the asymmetric cyanation of aldehydes. The homometallic heterogeneous catalyst was very active and selective, particularly at low loadings, highlighting the positive effect of confinement. As a result, the bimetallic Cu- and V-containing MOF was far less active, as the synergistic VO–VO pathway was blocked by the presence of the copper sites, resulting in less efficient unimolecular activation of the substrates. In the context of tandem catalysis, they proposed a similar strategy for the post-synthetic exchange of achiral ligands from Zr-based UiO-68 MOF with chiral salen complexes, bearing the same or different metal centers [59]. Consequently, a Mn-salen derived UiO-68 was obtained, which cannot be achieved by direct solvothermal synthesis. Then, from Mn-salen derived UiO-68, mixed salen-containing MOFs were obtained (Mn/Cr and Mn/V) by another solvent-assisted linker exchange procedure. These highly stable platforms were precisely characterized using NMR spectroscopy, ICP-OES, and Powder X-Ray Diffraction (PXRD). Excellent heterogeneous asymmetric activity of the newly formed UiO-68-(monometallic salen) MOFs was achieved depending on the nature of the trapped metal, such as epoxidation for the Mn-containing material and epoxide ring opening for Cr-one, for example. Finally, in the presence of the MOF UiO-68-(bimetallic Mn/Cr salen), a sequential tandem transformation (i.e. epoxidation followed by ring opening with anilines) was carried out to give the targeted amino alcohol in very high yield and almost complete enantioselectivity, with a low catalyst loading (0.5 mol%). This bimetallic catalyst was recycled after extraction of the products for at least 10 cycles without any loss of efficiency.
A variation in the structure of MOFs was also proposed by Ren, Jiang and their group when they heated a mixture of tetracarboxyphenyl-porphyrin and a Ni-salen complex in the presence of Cd(NO3)2 [60]. They obtained a porphyrin-salen based chiral MOF, with two different interpenetrated networks, one in which the porphyrin core contains Cd2+, the other being metal-free. Efficient cooperativity could be demonstrated using this MOF in catalytic asymmetric cyanosilylation (8 examples), due to the presence of the high Lewis acidic sites of the Cd2+ ion in the porphyrin ring. The close location to the enantiopure Ni-salen complex led to high values in terms of activity (up to 96% conversion) and enantioselectivity (up to 98% ee). Finally, we chose to highlight the immobilization of bulky Spinol-based chiral phosphoric acids in stable Zr-MOFs, and their use as asymmetric Brønsted acid catalysts to promote enantioselective multi-component tandem reactions [61]. The structure of the chiral organocatalysts was designed to protect them from coordinating with metal ions and to allow the formation of MOFs with different channel sizes. The corresponding heterogeneous catalysts were found to be more active than analogous homogeneous catalysts, with increased acidity in the two-component acetalization/Friedel–Crafts tandem reaction for the synthesis of enantioenriched 2,3-dihydroquinazolinones (11 examples, up to full conversion and enantioselectivity) or iso-Pictet–Spengler reaction of o-aminobenzylindole with trifluoromethylated ketones (7 examples, up to 98% yield and 96% ee). The catalyst was reused 10 times for the first transformation without losing its activity or enantioselectivity. A successful three-component tandem reaction using indole, aldehydes and p-toluenesulfonalide as substrates (Scheme 22) demonstrated interesting synergistic effects with the Lewis acid Zr(IV) sites.
Immobilization of a Spinol-based chiral phosphoric acid in Zr-MOFs for multi-component tandem reactions.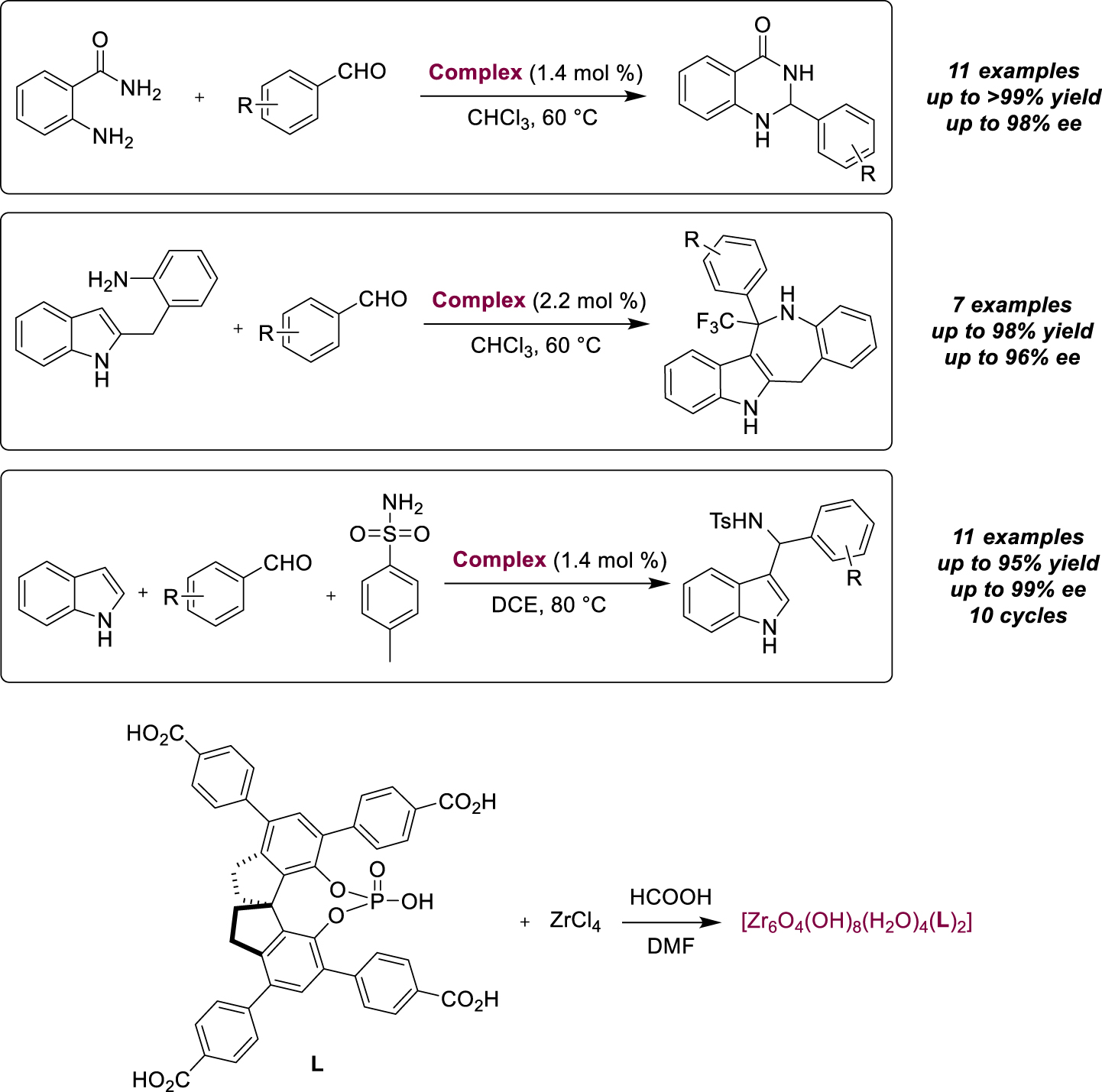
Similarly, the recent description of supramolecular coordination cages has also produced excellent results for asymmetric catalysis [62]. For example, various tetrahedral cages were prepared by mixing four clusters of Cp3Zr3 with six dicarboxylate ligands derived from metal-salen complexes. These new nanoreactors were described in detail and the authors succeeded in obtaining cages containing identical or different active catalytic sites [63]. Cages containing Mn-salen were used for the epoxidation of chromene derivatives with iodosylbenzene derivatives with enantioselectivity values higher than those obtained with the monomer. On the other hand, Cr-salen cages have been used successfully for the ring-opening of epoxides with aniline or trimethylsilylazide. Consequently, the mixed Cr/Mn-salen cage was tested in the sequential asymmetric epoxidation/ring-opening reaction of alkenes with TMSN3 (3 examples) or anilines (9 examples) and gave excellent results in terms of activity and enantioselectivity, outperforming those obtained using a 1/1 mixture of each cage containing either Cr-salen or Mn-salen as the active species, demonstrating the positive confining effect of a mixed cage. These few examples are only representative of a growing number of articles in this field, which show the value of these crystalline objects, which are perfectly defined and whose structure has been precisely targeted to carry out extremely varied asymmetric catalysis and asymmetric multicatalysis reactions, in highly efficient recycling procedures.
6. Ship in a bottle procedure
Finally, a last strategy based on non-covalent interactions to immobilize chiral catalysts is worth mentioning, namely the “ship in a bottle” strategy, which consists of trapping a homogeneous catalyst in a solid porous matrix, leaving the space for the entry of substrates and the exit of products. This procedure proved effective in immobilizing a chiral Co-salen complex inside the mesoporous cage of SBA-16, by controlling the size of the pore entrance, adapting the autoclaving time and the silylation process [64]. The corresponding heterogeneous chiral catalyst was successfully used for the asymmetric hydrolytic ring-opening of epichlorohydrin and propylene oxide with, in each case, values corresponding those obtained with the homogeneous counterpart. Furthermore, this catalyst was reused after simple filtration in ten recycles with propylene oxide as substrate, showing almost no loss in activity or selectivity, as a proof of the stability of the material and its ability to effectively trap active salen species. Another more recent example concerns the encapsulation of Mn-salen complexes in a one-step procedure for the synthesis of a highly porous MOF, namely NH2-MIL-101(Al), implying the linker, the Mn-salen complex and AlCl3 [65]. The corresponding crystalline material was characterized indicating the presence of a salen complex in one cage out of six. Although a lower conversion was obtained compared to the homogeneous catalyst, the enantioselectivity for the epoxidation of dihydronaphthalene remained unchanged at 70% ee, probably due to the soft encapsulation strategy where the catalyst structure remained unchanged, without influence of the MOFs walls. The MOF-cage thus assembled around the Mn-salen complex was tested for four recycles with a slight decrease in activity and no loss of selectivity. Further analytical studies of the solid catalyst and filtrate were carried out, showing an unchanged structure and no leaching, demonstrating the stability and truly heterogeneous nature of the catalyst. A final example involves the entrapment of a chiral Co-salen catalyst in IRMOF-3 nanocages by adsorption. As this support is derived from tetranuclear Zn4O clusters linked to 1,4-dicarboxylic benzene linkers with a functional amino group, subsequent acylation of the free amino groups via the post-synthetic modification technique enabled the catalytic sites to be trapped by reducing pores size and, therefore, access [66]. The synthesis of enantioenriched cyclic carbonates from epoxides and CO2 was used as a test reaction resulting in a product with slightly reduced activity compared with the homogeneous case, probably due to reduced diffusion. The control experiments nevertheless proved the existence of heterogeneous activity inside the pores, with the catalyst easily recovered and effectively reused over four repeated runs.
7. Conclusion and outlook
The immobilization of enantiopure catalysts, whether organic species or organometallic complexes, via non-covalent interactions on various support is constantly evolving. The use of weak and reversible interactions could lead to leaching of the catalyst and hinder, for example, efficient recycling or promote metal contamination of the target product. While this may still be true for certain methods of immobilization by electrostatic interactions on silica-type supports, partly because of their polar nature, many other solutions have been found, for example by adapting the solvent so that the effective recycling number reaches very high values. More recently, π-interactions with carbon supports have also been exploited in the field of asymmetric catalysis, enabling simple procedures to be implemented since no modification of the support is required. The conservation of the activity and enantioselectivity of the immobilized catalyst was described, as well as numerous effective recycles, right up to implementation in flow procedures as proof of the robustness of this heterogenization process. Future developments of this method should involve the co-immobilization of different (chiral) active species on the same support in order to promote ambitious asymmetric multicatalytic reactions. The formation of charge transfer complexes for the recycling of chiral catalysts is rarer, but has also proved its worth, either by acting in the homogeneous phase and being easily recovered by the addition of an apolar solvent before filtration, or by direct filtration when the CTC is formed directly with a species anchored on a support. Examples of numerous recycling processes are also described here as an interesting method for recovering and reusing, not only the catalyst but also the support, through easy exchanges of active species thanks to non-covalent interactions. These immobilization procedures have also been used in multi-substrates reactions, as well as in multi-reaction procedures proving both their applicability and their robustness. The use of MOFs for supported asymmetric catalysis is growing rapidly; in fact, today’s perfect mastery of the synthesis of these crystalline materials, whose structure and porosity are controlled, means that we can decide on the exact location of the catalytic sites, spacing them out or bringing them closer together as required, depending on the desired reactivity, whether individual or cooperative. These perfectly designed nanoreactors offer size selectivity, multiple recycling without metal contamination and asymmetric multicatalysis. The field of supported asymmetric catalysis, involving the use of non-covalent bonds, is still wide open to creativity, to easily carry out numerous transformations using the support, not only for the purposes of economy and recycling, but also to control the placement of numerous active catalytic sites for the discovery of increasingly complex reactivities.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgments
We are grateful to the CNRS, the Université Paris-Saclay for their support and to the Agence Nationale de la Recherche (ANR-22-CE07-0A40-01, PhD financial support to LI). For the purpose of Open Access, a CC-BY public copyright licence has been applied by the authors to the present document and will be applied to all subsequent versions up to the Author Accepted Manuscript arising from this submission.