

1 Introduction
Naturally occurring amino acids are of particular interest in the field of biocompatible surfactants. Diacylglycerol amino acid conjugates constitute a novel class of speciality biocompatible surfactants, which can be considered analogues of partial glycerides and phospholipids. They consist of two aliphatic chains and one polar head, i.e. the amino acid, linked together through a glycerol moiety. In this sense, our group have recently synthesized diacylglycerol amino acid-based surfactants [1,3]. Preliminary results on physicochemical and biological properties show that these novel compounds combine the advantages of both partial glycerides and lipoamino acids. We have observed that they possess antimicrobial activity, like long chain Nα-acyl amino acid derivatives [1,2], and form lamellar phases and vesicles [4,5], characteristic of partial glycerides and phospholipids.
In this work an enzymatic procedure for the synthesis of dilauroylated arginine glyceride conjugated was used. The strategy devised for their synthesis consists of two steps. The first involves the enzymatic preparation of the arginine glyceryl ester derivative [rac-1-O-(Nα-acetyl-L-arginine) glycerol] [6]. In the second step, the enzymatic acylation of the hydroxy groups of the arginine glyceryl ester by fatty acids is performed [3].
As in conventional glycerides, spontaneous intramolecular acyl-migration reactions are observed for the lauroyl and aminoacyl moieties of the glycerol backbone [2] and both possible regioisomers are obtained (Fig. 1). This spontaneous reaction often obscures the data from lipid modification reactions and it makes it difficult to precisely determine the compound distribution of finished products. It is known that partial glycerides dissolved in organic solvents undergo acyl migration, whose rate depends on the solvent, temperature and ion concentration [7]. In the solid state, this process is slower, but the acyl migration has been still observed [8].
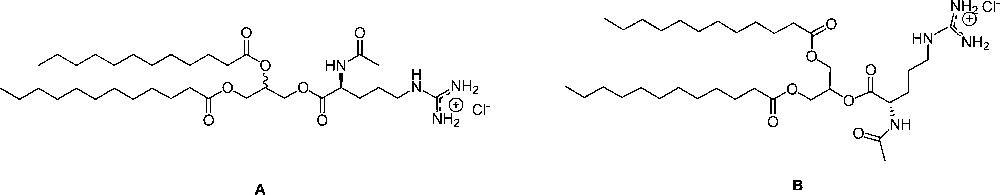
The chemical structure for regioisomers dilauroylated products: 1,2 di-lauroyl-rac-glycero-3-(Nα-acetyl-L-arginine) (A) and 1,3-di-lauroyl-glycero-2-(Nα-acetyl-L-arginine) (B) pure or their mixtures used in this study.
The influence of the presence of both regioisomers on the physicochemical properties of these new compounds was investigated. To this end, the phase behaviour of pure and regioisomeric mixtures of these compounds were examined by differential scanning calorimetry, small-angle X-ray scattering and by polarized light microscopy.
2 Experimental
2.1 Materials
Papain (EC 3.4.22.2.) from papaya latex crude powder [1.7 U mg–1 of solid, one unit (U) will hydrolyse 1.0 mmol of Nα-benzoyl-L-arginine ethyl ester hydrochloride per minute (BAEE) at pH 6.2 at 25 °C] was obtained from Fluka. Bacillus subtilis protease (AlcalaseTM) (EC 3.4.21.62) (0.6 U mg–1), Candida antarctica lipase (Novozym 435TM) (EC 3.1.1.3.) (7000 PLU g–1, PLU: propyl laurate units) and Rhizomucor miehei lipase (Lipozyme IMTM) (EC 3.1.1.3.) (5–6 BAUN g–1, BAUN: Batch Acidolysis Units Novo) were a generous gift from Novo Nordisk A/S.
Nα-acetyl-L-arginine [which was converted to Nα-acetyl-L-arginine hydrochloride by treatment with HCl] was obtained from Novabiochem. Glycerol, 1,4-dithio-DL-threitol (DTT) and CeliteTM (type 545, particle size 26 μm; mean pore diameter 17 00 nm; specific surface area, BET method, 2.19 m2 g–1) were obtained from Fluka. Solvent and other chemicals used in this work were of analytical grade.
2.2 Methods
2.2.1 Enzyme adsorption
The general procedure for the deposition of papain onto solid support materials was the following. Papain (100 mg) and DTT (50 mg) were dissolved in 0.1 M boric–borate pH 8.2 buffer and mixed thoroughly with CeliteTM (1 g). The mixture was then evaporated under vacuum overnight.
2.2.2 HPLC analysis
The amount of reactants and products in the enzymatic reactions was measured by HPLC analysis. HPLC analyses were performed on a Merck-Hitachi Lichrograph system using a Lichrocart 250-4 HPLC cartridge, 250 × 4 mm filled with Lichrosphere ® 100, RP-18, μm or Lichrosphere® 100 CN (Propylcyano) (Merck). In the synthesis of rac-1-O-(Nα-Ac-L-arginyl) glycerol hydrochloride, samples (8 μm) were withdrawn from the reaction medium, mixed with acetic acid (8 μm) to stop any further enzymatic reaction, evaporated under vacuum, and dissolved in HPLC eluent before analysis. Quantitative analysis was performed from peak areas by means of the external standard method. Solvent system: solvent [A]: 0.1% (v/v) trifluoroacetic acid (TFA) in water, solvent [B]: 0.085% (v/v) TFA in water/CH3CN 1:4, isocratic elution 0% [B]. K′-values: Ac-Arg-OH (2.6), Ac-Arg-OGl (5.2).
In the synthesis of 1,2 (1,3)-di-O-lauroyl-rac-glycero-3(2)-O-(Nα-acetyl-L-arginine) products, samples (5–10 μm) were withdrawn from the reaction medium at different times. The samples were dissolved with a solution of acetonitrile/water/acetic acid 16:3:1 (2 ml) to stop any further enzymatic reaction, centrifuged to separate the solid particles, and the supernatant was analysed directly by HPLC on the 100 CN (Propylcyano) column. This provided the qualitative and quantitative analysis of the lauric acid, mono- and dilauroylated products. Solvent system: solvent [C]: aq. Triethylamine-phosphoric acid buffer. pH 6.0 (TEAP pH 6.0) in H2O, solvent [D]:TEAP (pH 6.0)/CH3CN 1:4, gradient elution from 35 to 100% [D] in 26 min; K′-values: lauric acid (2.6), monolauroylated (6.2) and dilauroylated (11.6) products. After that, an aliquot of the supernatant obtained previously (500 μm) and the solid formed was centrifuged. The supernatant was evaporated under vacuum to dryness, partly dissolved in water (500 μm), and the residue was centrifuged again. The supernatant was analysed directly by HPLC on the C18 column, allowing the quantification of rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride. In all cases, the flow rate was 1 ml min–1, with UV detection at 215 nm.
2.2.3 Enzymatic synthesis
2.2.3.1 Synthesis of rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride
The Nα-acetyl-L-arginine hydrochloride (48.6 mmol) was dissolved in glycerol (100 ml) at 50 °C, containing H3BO3/Na3BO3 0.2 M pH 8.2 buffer (10% v/v) and the scavenger DTT (2.0 g). The reaction was initiated by the addition of the enzyme papain (2.8 g) and monitored by HPLC. At the end, the reaction mixture was diluted with a mixture of MeOH/acetic acid 4:1 to stop any further enzymatic reaction. The mixture was filtrated through CeliteTM and the filtrate obtained was evaporated under vacuum. Then, the oily residue obtained was dissolved with plain, deionised water and purified by ion-exchange chromatography. Physical and spectroscopic data matches as previously published in [6].
2.2.3.2 Synthesis of 1,2 (1,3)-di-O-lauroyl-rac-glycero-3(2)-O-(Nα-acetyl-L-arginine) derivatives
The rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride (11.0 mmol) was dissolved in aqueous phosphate 1.0 M pH 7.0 (44.0 ml). To this solution was added lauric acid (90.0 mmol) and, after stirring, the mixture was freeze-dried during 48 h. To the solid obtained was added water [4% w/w with respect to the rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride] and the reactor was placed at 65 °C. The reaction was initiated by the addition of the enzyme (2.2 g) and monitored by HPLC. At the end of the reaction, the mixture was diluted with a solution of CH3CN/water/acetic acid 16:3:1 to stop any further enzymatic reaction and to dissolve the solid particles of product and substrates. The resulting mixture was filtered through CeliteTM-545.The filtrate was concentrated until both 1,2 (1,3)-di-O-lauroyl-rac-glycero-3 (2)-O-(Nα-acetyl-L-arginine) derivatives and lauric acid precipitated out and the residues were pooled. The residues were dried under vacuum and purified by preparative HPLC. Physical and spectroscopic data matches as previously published in [3].
2.2.4 Polarized light microscopy
A polarizing microscope (Reichert Polyvar® 2) equipped with a heating stage was used to observe the texture of samples during heating process. Samples were heated from 20 to 100 °C. For the image capture at different temperatures, a video camera and a PC with a software Leica IM 500 was used.
2.2.5 Differential scanning calorimetry (DSC)
DSC measurements were carried out using a 821e DSC (Mettler Toledo) equipped with a cryostat cooling system. Data were treated, mathematically using the STAR Software (Mettler Toledo). A flow rate of 50 ml min–1 of nitrogen was used in the DSC cell. Samples (3–4 μg) of the dry lipids were scanned using hermetically sealed aluminium pans of 40 μl volume (Mettler Toledo). Indium as standard was used to calibrate the DSC temperature scale and the enthalpic response.
2.2.6 X-ray scattering (SAXS)
Small angle X-ray scattering (SAXS) measurements were performed using the methodology described in Plaza et al. [9]. The range of the scattering vector, defined as q = 4 π sin(θ)/λ,where λ is the wavelength and 2θ is the scattering angle, was from 0.01 to 0.6 Å–1 (corresponding distances between 600 and 11 Å, d = 2 π/q). X-ray spacing is generally accurate to ±0.5 Å and temperatures of the X-ray specimen stage were controlled to better than ±0.5 °C.
3 Results and discussion
3.1 Enzymatic synthesis
The enzymatic synthesis of dilauroylated arginine glyceride conjugated derivatives was achieved following a two-step procedure. The first step involves the enzymatic preparation of the arginine glyceryl ester derivative [rac-1-O-(Nα-acetyl-L-arginine) glycerol]. In the second step, the lipase-catalysed acylation of the hydroxy groups of the arginine glyceryl ester by fatty acids is performed (Fig. 2).

Reaction pathway for the synthesis of diacylglycerol amino acid conjugates.
3.1.1 Synthesis of rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride
The synthesis of rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride involves the esterification of one of the primary hydroxy groups of glycerol by the α carboxy group of the amino acid arginine.
The enzymatic acylations were carried out in solvent-free media, considering both the environmental standpoint and the recent trends in biocatalysed reactions [10]. Hence, glycerol was used as reagent and solvent. The reactions were performed in glycerol containing 10% (v/v) buffer concentration at 50 °C.
We were interested in investigating the ability of industrial hydrolases such as Novozym 435TM, AlcalaseTM and Papain as putative biocatalyst for the esterification of Ac-Arg-OHHCl with glycerol (Table 1). For papain, a 74% yield was achieved when it was used as soluble enzyme. For AlcalaseTM and Novozym 435TM, yields of 79 and 90% were achieved. An important advantage of AlcalaseTM and Novozym 435TM is that scavengers, used with papain to prevent the oxidation of its active site, can be avoided. The product was purified by cation-exchange chromatography [6]. The corresponding isolated yields are shown in Table 1.
Hydrolase-catalysed synthesis of rac-1-O-(Nα-Ac-L-arginyl) glycerol hydrochloride. The enzymes AlcalaseTM and Novozym 435TM were used as powder or liquid (straight from the bottle). Papain was assayed as powder and deposited onto CeliteTM 545
| Enzyme | Configuration | Reaction yield (%) | Isolated yield (g) (% of theor.) |
| Papain | Deposited | 22 | |
| Free | 74 | 13 (67) | |
| AlcalaseTM | Free | 79 | 8 (59) |
| Novozym 435TM | Deposited | 90 | 15 (80) |
1H-NMR analysis of the product reveals the presence of 2-O-regioisomer [2-O-(Nα-acetyl-L-arginyl) glycerol] in ~10%. In agreement with the literature data [11], this result indicated that the enzymes exhibited a preference towards acylations of one of the primary hydroxy groups in the glycerol molecule (i.e. alcohol groups in C1 or C3 positions). The 2-regioisomer, although it could be produced enzymatically to some extent, may be formed preferentially by simple 3(or 1)→2 spontaneous acyl migration similarly to that observed in mono- and diglycerides [7] and monoesters of Boc-Ala-OH with propane-1,2-diol [11].
It was ascertained that the enzymes used could not distinguish between the two enantiotopic hydroxymethyls of glycerol. The presence of both diastereoisomers was detected and quantified (approximately 50% of each) by 1H-NMR.
3.1.2 Synthesis of 1,2 (1,3)-di-O-lauroyl-rac-glycero-3(2)-O-(Nα-acetyl-L-arginine) derivatives
The synthesis of 1,2 (1,3)-di-O-lauroyl-rac-glycero-3(2)-O-(Nα-acetyl-L-arginine) derivatives consists of the lipase-catalysed esterification of two hydroxy groups of the rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride with lauric acid. To this end, both 1,3-regioselective lipase (Novozym 435TM) and non-selective lipase (Lipozyme IMTM) were selected, taking advantage of the spontaneous intramolecular acyl-migration reaction that occurs in partial glycerides [7,12].
The enzymatic acylations were carried out in solvent-free media. The melted substrate approach was used to achieve intimate contact of the solid substrate mixture [13,14]. The reactions were performed at temperatures higher than the melting point of the lauric acid ( 44 °C). The melted lauric acid acted as reagent and solvent.
Novozym 435TM and Lipozyme IMTM were versatile catalyst for this reaction. In many instances, the reaction performance of both lipases was similar. The use of Lipozyme IMTM favoured the yield of dilauroylated product due to the effect of the immobilization support on the rate of intramolecular acyl migration [15]. Preliminary studies showed that the buffer salts concentration had a strong effect on reaction performance with both lipases. Using Novozym 435TM, no reaction was observed in the absence of buffer salts, whereas Lipozyme IMTM gave 55% conversion during the esterification reaction in the absence of salts. The reactions needed 560 mg mmol–1 of buffer salts, a temperature of 65 °C and a ratio of lauric acid to Ac-Arg-OG of 8:1 to give sufficient substrate conversion for the preparation of dilauroylated conjugated in gram quantities (Table 2).
Effect of the molar ratio, temperature and biocatalyst on the maximum product reaction yields
| Molar Ratio | T (°C) | Enzyme | Reaction yield (%) | Isolated yield (g) (% of theor.) |
| 4.1 | 55 | Novozym 435TM | 8 | |
| Lipozyme IMTM | 33 | |||
| 65 | Novozym 435TM | 59 | ||
| Lipozyme IMTM | 54 | |||
| 8:1 | 55 | Novozym 435TM | 19 | |
| Lipozyme IMTM | 60 | |||
| 65 | Novozym 435TM | 49 | ||
| Lipozyme IMTM | 74 | 1.7 (22) |
HPLC analysis of the reactions samples showed that the monolauroylated products gave broad peaks, whereas for the dilauroylated products, two signals were distinguished although not baseline separated (less than 0.2 min of retention time). Analysis of the samples by liquid–liquid mass spectroscopy (LC/MS) demonstrated that this behaviour was caused by the different isomers of the mono and dilauroylated products. The structure of these isomers was ascertained unequivocally by mono- and bidimensional NMR spectroscopic techniques [3]. The percentage of 1,3-dilauroylated regiosomer was 30%. Moreover, the dilauroylated products were equimolar mixtures of diastereoisomers. It seems that the used lipases were not selective between the two diastereoisomers of the starting rac-1-O-(Nα-acetyl-L-arginyl) glycerol hydrochloride and to those generated during the monolauroylation reaction. The purification of both 1,2 (1,3)-di-O-lauroyl-rac-glycero-3(2)-O-(Nα-acetyl-L-arginine) derivatives were carried out by silica acid flash chromatography [4].
Using differential scanning calorimetry (DSC), small-angle X-ray diffraction (SAXS) and polarized light microscopy the phase behaviour of 1,2-di-lauroyl-rac-glycero-3-(Nα-acetyl-L-arginine) (compound 1) and its mixtures with the 1,3-di-lauroyl-glycero-2-(Nα-acetyl-L-arginine) derivative was investigated (compounds 2 and 3, with a molar percent of 1,3-di-lauroyl-glycero-2-(Nα-acetyl-L-arginine) of 30 and 50% , respectively).
3.2 Qualitative phase behaviour
The compounds, lyophilised from aqueous solutions, were white solid highly hygroscopic. At room temperature, the samples exhibited birefringent texture under the polarization microscope, indicating the presence of a crystalline structure. On initial heating of the solids, softening was observed, the exact temperature being dependent on the particular compound. The ‘softened’ materials are birefringent, having ill-defined ‘cloudy’ textures (Fig. 3A). At higher temperatures, these phases transform to a classical smectic (or lamellar) phase (Fig. 3B), which subsequently melts at temperatures around 90 °C. On cooling the isotropic liquid and with a subsequent reheating, the smectic phases were formed at temperatures close to 20 °C.

Representative optical polarizing micrographs displayed by the compound 1 upon the first heating: smectic phase at 45 °C (A); positive units and oily streaks in the smectic phase at 70 °C (B).
3.3 DSC studies
The thermotropic mesophase formation of the compounds synthesized was studied in details by DSC techniques. Measurements were conducted at heating and cooling rate of 5 °C min–1 over a temperature range between –30 and 140 °C. Representative thermograms of these compounds are shown in Fig. 4.
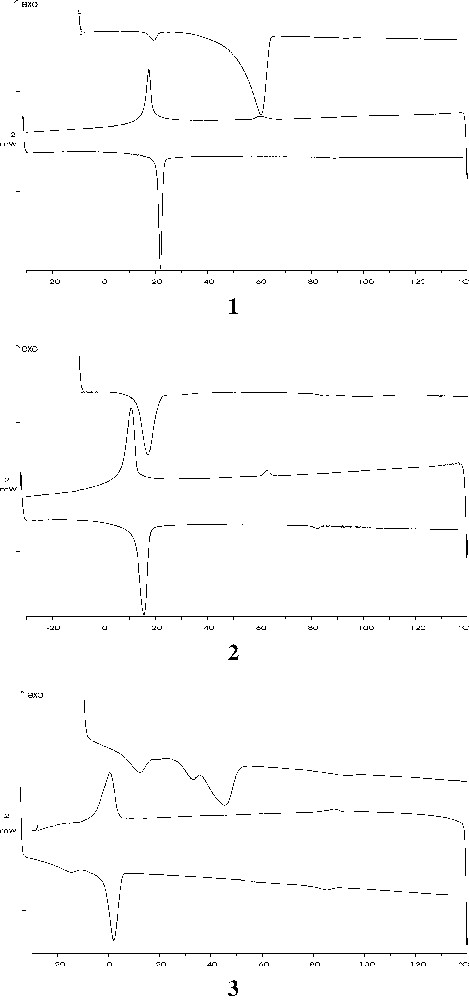
DSC thermograms of compounds 1, 2 and 3. The experiments were performed for a sequence of heating, cooling and reheating scans.
The data show that upon initial heating of the crystalline compounds 1 and 3, there was a large endothermic transition at temperatures well below those at which the isotropic liquid was formed. The appearance of this very energetic endothermic transition is probably attributable to the formation of a smectic liquid-crystal mesophase (see the X-ray diffraction section). Similar transition has been observed with lipids and glycoside detergents [16]. Under cooling, the isotropic liquid converts back to the mesophase. This transition was not a simple reversal of the solid-to-liquid-crystal transition, as observed on the initial heating of these compounds, since it occurred at a lower temperature and was considerably less energetic. Reheating of the sample, however, showed that this second solid/liquid-crystal transition is reversible. Results indicate that the thermotropic interconversions between the isotropic liquid, the mesophase and the solid phase ‘crystallized’ from the melt are reversible. In all the compounds, a feebly endotherm appears at high temperature (i.e. temperatures above 90 °C), which was proved by optical microscopy to be the clearing point. This behaviour clearly confirms the occurrence of the mesophases observed by optical microscopy.
In the isomeric mixtures (compounds 2 and 3), the presence of regioisomer 1,3- perturbs the structure of the gel phase by decreasing the temperature of the gel/liquid-crystal transition (Fig. 4). This transition temperature shows a linear decrease, as the mole percent of 1,3-regioisomer is increased, from 17 °C for compound 1 to 0.5 °C for the equimolar mixture (compound 3).
To further investigate the structure of the phases formed by these compounds, SAXS measurements were performed during heating and cooling modes in the range temperatures between 0 and 70 °C, where the mesophase transitions were observed with the DSC experiments.
3.4 X-ray studies
SAXS measurements of compounds 1, 2 and 3 confirm the pattern of thermal events observed by DSC and show that the phases on either side of the DSC transitions have lamellar characteristics. Two or more order peaks were observed and, in any case, at least two peaks were used for fitting. Using the Bragg equation, the repeat period d of the lamellar phase was determined from the reciprocal spacing of the maximums of SAXS peaks.
All d spacing are smaller than the twice length of a fully extended C12 chain (17 Å) [17]; hence, a layer or bilayer structure appears likely. The larger bilayer thickness of compound 1 compared with compounds 2 and 3 is attributable to full extended, all-trans alkyl chains. For compounds 2 and 3, the difference in bilayer thickness might be associated with a tilting of the hydrocarbons chains away from the normal bilayer. For the compounds 2 and 3, the calculated tilting from the normal bilayer is 14 and 38°, respectively. This more ordered structure for compound 1 compared with compounds 2 and 3 agrees with the smaller value observed in the peak width for compound 1 as compared to compounds 2 and 3.
Upon passage through the main transition, all compounds undergo a reduction in the lamellar d spacing that corresponds to a 10% decrease in the chain length. Assuming a molecular model with temperature independent head-group structure, this difference in bilayer thickness might be associated with the introduction of one kink in the chain. At temperatures close to 70 °C, no difference is observed in the bilayer thickness of these three preparations. In all cases, the reduction of chain length is compatible with the introduction of three kinks in the chain, i.e. the molten chains [17].
Acknowledgements
This work was supported by the Spanish CICYT, ref. PPQ2000-1687-CO2-01. We are grateful to M. Jaime Caelles and M. Josep Carilla for their technical assistance in the SAXS and DSC experiments, respectively.