

1 Introduction
The development of new chiral heterogeneous catalysts to promote enantioselective reactions is a field of growing interest due to the inherent advantages of heterogeneous catalysis in the industrial preparation of fine chemicals and specialities [1]. Transition and lanthanide elements complexed with C2-symmetric pyridine-bis(oxazoline) (pybox) ligands have been used successfully in many asymmetric organic reactions [2–5]. However, heavy metal-based catalysts have been found to be extremely toxic [6]. The immobilization of this very important kind of catalysts could be the first step for their use within the green chemistry philosophy. In this communication, we describe very simple strategies to immobilize pybox by formation of a covalent bond between the pyridine ring and the insoluble support.
2 Results and discussion
Starting from a 4-bromopybox 1 [4], two 4-vinyl and 4-(4-formylfenoxy)pybox derivatives 2 and 3 were prepared (Fig. 1). Vinyl derivative 2 was used in different block co-polimerization with styrene and divinylbenzene, using a porogen and AIBN for the preparation of monolithic resins [7]. Three different polymers (P1–P3) were obtained from vinylpybox 2 by changing the degree of cross-linking and the porogen (Table 1). In all cases, the pybox ligand was incorporated into the polymer with high yield (0.40–0.44 mmol g–1), as shown by the elemental analysis data.
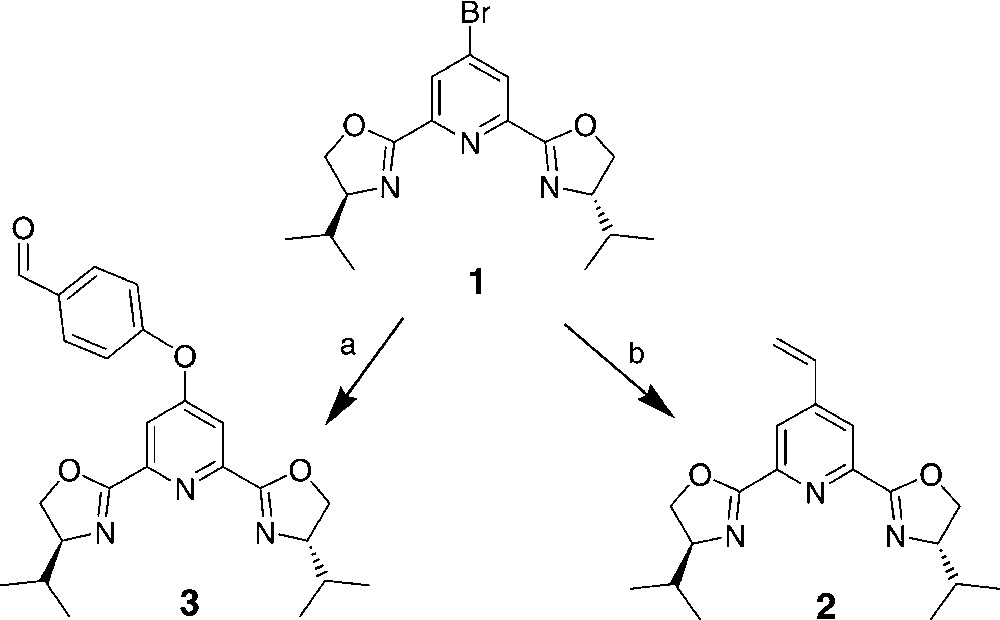
Preparation of the 4-vinyl and 4-(4-formylfenoxy)pybox derivatives 2 and 3 starting from a 4-bromopybox 1.
Catalysts used in this work
| Catalyst | Polimerization mixture (%) | porogen | pyboxa | Rub | ||
| pybox (7) | styrene | DVB | (mmol g–1) | |||
| P1 | 7 | 42 | 51 | toluene | 0.44 | 0.27 |
| P2 | 7 | 63 | 30 | tol/dodecc | 0.43 | 0.26 |
| P3 | 7 | 42 | 51 | tol/dodecc | 0.41 | 0.14 |
| S1 | — | — | — | — | 0.15 | 0.10 |
| S2 | — | — | — | — | 0.42 | 0.24 |
On the other hand, the formyl derivative 3 was immobilized using an imine bond formation on silica gel. Merck 60 silica, with surface area of 500 m2 g–1 and pore diameter of 60 Å, was used as a support. Silica S1 was prepared from Merck 60 by sequential treatments with hexamethyldisilazane, 3-aminopropyltriethoxysilane and the formyl-pybox 3. The resulting solid showed a ligand content of 0.15 mmol g–1 (Table 1). However, the content of the ligand increased to 0.42 mmol g–1 when silica gel was firstly treated with the linker 3-aminopropyltriethoxysilane, and subsequently with the formyl derivative 3 and then end-capped with hexamethyldisilazane (S2 in Table 1).
These immobilized ligands were transformed into ruthenium catalysts by treatment with [RuCl2(p-cymene)]2 (Fig. 2). Ru-functionalization was in general low, which seems to indicate that a significant proportion of the ligand is situated in inaccessible parts of the solid. In the case of polymers, the accessibility is controlled by the porogen (toluene better than toluene/dodecanol mixture) and the cross-linking degree. On silica, the surface concentration of groups seems to be too high to accommodate bulkier groups in the next step. The comparison of the elemental analysis of S1, S2 and silica-linker intermediates shows that 50% of the amino groups of the silica-linker remain free in S1 and S2 after the treatment with pybox. In a similar way, only 60–70% of the pybox is able to form the Ru-complex.
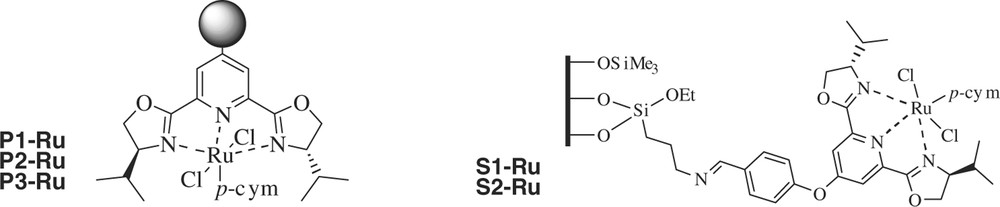
Transformation of immobilized ligands into ruthenium catalysts by treatment with [RuCl2(p-cymene)]2.
The asymmetric cyclopropanation of styrene with ethyl diazoacetate was used as the benchmark reaction test. In all cases, the heterogeneous catalyst was filtered off and an additional quantity of ethyl diazoacetate was added to the solution to confirm the heterogeneous character of the catalyst [8].
As shown in Table 2, the polymerised pybox P1–Ru showed behaviour in trans/cis selectivity and trans enantiomeric excess close to that of the homogeneous catalyst H–Ru. The use of dodecanol in the porogenic mixture was detrimental for both selectivities (P1–Ru vs P3–Ru), in agreement with the reduced accessibility evidenced by the lower Ru functionalization. The improvement in accessibility obtained with a lower degree of cross-linking (P2–Ru vs P3–Ru) was also accompanied by an improvement in selectivities. The best catalysts were reused twice, with similar efficiency in the first recycle, but a marked decrease in selectivities, as well as in activity in the case of P1–Ru (Table 2). The coordination of by-products may be responsible for this deactivation, with a partial de-coordination of Ru from the chiral ligand, which can act as a bidentate ligand with the corresponding decrease in enantioselectivity.
Results obtained in the cyclopropanation reaction
| Catalystb | Run | Yield (%)c | trans/cisc | % ee trans | % ee cisd |
| H–Rue | 1 | 34 | 90/10 | 88 | 70 |
| P1–Ru | 1 | 31 | 85/15 | 85 | 41 |
| 2 | 28 | 84/16 | 84 | 40 | |
| 3 | 11 | 75/25 | 45 | 20 | |
| P2–Ru | 1 | 32 | 78/22 | 76 | 41 |
| 2 | 35 | 85/15 | 75 | 42 | |
| 3 | 28 | 75/25 | 44 | 18 | |
| P3–Ru | 1 | 26 | 77/23 | 54 | 18 |
| 2 | 39 | 72/28 | 30 | 13 | |
| S1–Ru | 1 | 15 | 82/18 | 64 | 34 |
| S2–Ru | 1 | 35 | 73/27 | 38 | 12 |
The performance of the silica-immobilized catalysts was highly dependent on the immobilization method. When the pybox ligand was immobilized before end-capping (S2–Ru), the catalyst was as active as the polymeric ones, but the enantioselectivities were clearly lower. On the contrary, the end-capping with hexamethyldisilazane prior to other functionalizations (S1–Ru) led to a catalyst with low activity but moderate selectivities, comparable to those obtained from polymers P2–Ru and P3–Ru.
Molecular modelling studies were carried out in an attempt to explain the special features observed in functionalization and performance of the silica-supported catalysts. The semi-functionalization of the amino linker seems to be a consequence of the large section of the pybox nucleus together with dynamic aspects. These factors allow the pybox to shield the remaining amino groups (Fig. 3), preventing their functionalization. A similar shielding effect between pybox moieties might also be responsible for the incomplete complexation of immobilized pybox with ruthenium. The reduced enantioselectivity is much more difficult to explain and points to a contribution of non-enantioselective cyclopropanation, which requires the formation of non-chiral active sites on the solid. At first sight, the presence of free amino groups could account for the formation of this type of sites. However, the problems of accessibility to these amino groups seem to discard this hypothesis. Taking into account this idea, a possible explanation would be the participation in the ruthenium complexation of the free amino groups in the neighbourhood of pybox. The effect might be the deactivation of the site due to the coordination of an additional ligand or the partial de-coordination of pybox, which could act as a bi-dentate ligand, acting the amino group as the third coordinating group. The same role might be played by a close pybox ligand, due to their high surface density. In addition, the possibility of ruthenium complexation by free silanol groups of the support cannot be discarded.
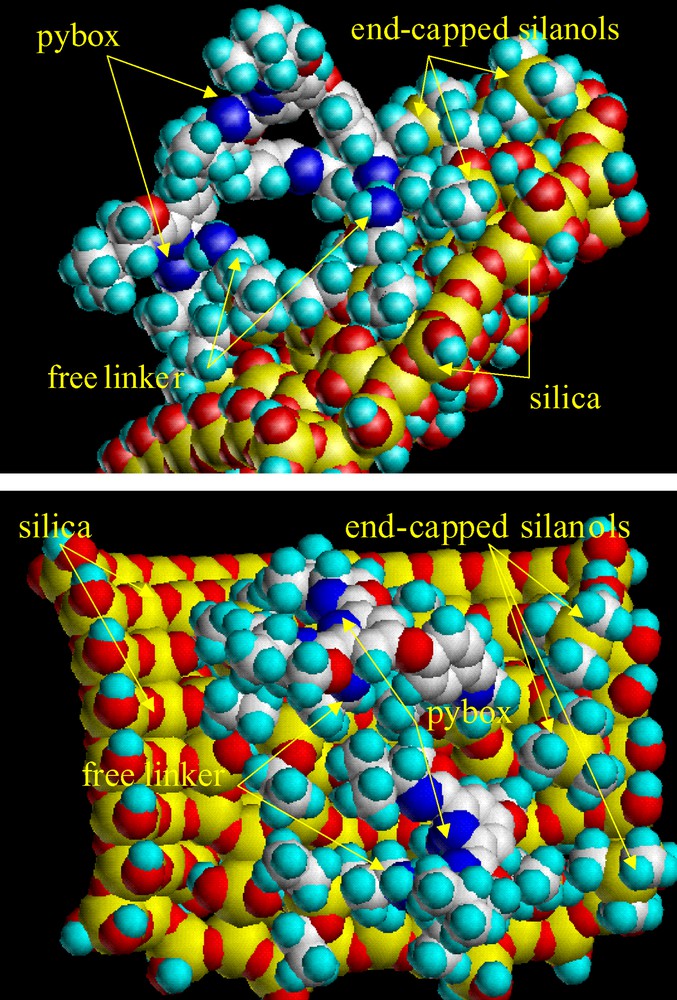
Snapshot from a Tripos dynamic simulation [9] of a simplified S1 silica, and subsequently minimised using MM2 force field [10]. Atom colours in the lateral (up) and azimuthal (down) views are as usual.
3 Experimental
All the reactions were carried out under an atmosphere of dry nitrogen using flame-dried glassware and freshly distilled dry solvents. Routine monitoring of reactions was performed using Alugram SilG/UV254 (0.20 mm). All the chromatographic separations were performed using silica gel (Merck 60 230–400 mesh). 1H NMR spectra were recorded on a Varian 200 MHz spectrometer with TMS as internal standard. Chemical shifts are reported in ppm and coupling constants in Hz. IR spectra of products 1–3 were recorded on an Avatar 360 FT–IR spectrophotometer. Self-supported wafers of the polymers were evacuated (< 10–4 Torr) at 50 °C and transmission FTIR spectra were taken with a Mattson Genesis Series FTIR. Ruthenium analyses were carried out by plasma emission spectroscopy on a Perkin-Elmer Plasma 40 emission spectrometer. Elemental analyses were carried out on a Perkin-Elmer 2400 elemental analyser. Results of the cyclopropanation reactions were determined by gas chromatography on a Hewlett-Packard 5890II with FID detector; helium as carrier gas, 20 p.s.i.; injector temperature: 230 °C; detector temperature: 250 °C.
3.1 (S,S)-4-(4-formyl)phenoxy-2,6-bis(4-isopropyl-2-oxazolin-2-yl)pyridine (3)
To a stirred solution of potassium carbonate (186 mg, 1.35 mmol) in DMF (1 ml) at room temperature, p-hydroxybenzaldehyde (165 mg, 1.35 mmol) was added. The resulting mixture was stirred at room temperature for 30 min. Then (S,S)-4-bromo-2,6-bis(4-isopropyl-2-oxazolin-2-yl)pyridine 1 (380 mg, 1.00 mmol) was added. The mixture was heated at 95 °C for 24 h. Then the resulting mixture is allowed to cool, poured onto ice water (40 ml) and stirred during 15 min. The resulting solid was collected by filtration and dissolved in ethyl acetate (50 ml) and washed with water (3 × 50 ml) and aqueous sodium hydroxide (0.25 M, 3 × 50 ml). The organic layer was dried over MgSO4 and then distilled under reduced pressure. The solid residue was recrystallized in hexane/ethyl acetate, yielding 231 mg (55%) of 3.
1H RMN (CDCl3, δ ppm): 9.99 (s, 1H); 7.95 (d, J = 8.8 Hz, 2H); 7.80 (s, 2H); 7.20 (d, J = 8.8Hz, 2H); 4.54–4.46 (m, 2H); 4.20–4.02 (m, 4H); 1.87–1.74(m, 2H); 0.99 (d, J = 6.6Hz , 6H); 0.88 (d, J = 6.6Hz, 6H).
IR (KBr, cm–1): 2958, 1695, 1644, 1581, 1397, 1230. MS m/z: 421, 406, 378, 351, 336, 307, 293, 263, 209. Melting point: 114–115 °C.
Elemental analysis: Calcd for C24H27N3O4: C, 68.41; H, 6.41; N, 9.98. Found: C, 68.47; H, 6.37; N, 9.84.
3.2 S1–Ru
3.2.1 End-capping
To a stirred suspension of 1.00 g of activated silica gel (Merck 60 Art. No. 109385, 0.040–0.063 mm, heated one night at 140 °C under vacuum) in anhydrous toluene (15 ml) at room temperature (r.t.), 1,1,1,3,3,3-hexamethyldisilazane (0.70 ml, 3.3 mmol) was added dropwise. The resulting mixture was refluxed for 1 h and then cooled at r.t. The solid was collected by filtration and subsequently washed with toluene (100 ml), methylene chloride (100 ml), ethanol (100 ml) and diethyl ether (100 ml) once each one. The solid was then dried at 50 °C under vacuum overnight with phosphorous pentoxide.
Elemental analysis. Found: C, 4.99; H, 1.17. Equivalent to 1.39 mmol of the trimethylsilyl ligand (referred to carbon) per gram of silica gel (end-capped silanols 1.39 mmol g–1).
3.2.2 Grafting of the amino spacer
To a stirred suspension of freshly prepared end-capped silica gel (1.00 g) in anhydrous toluene (15 ml) at room temperature, 3-aminopropyltriethoxysilane (0.25 ml, 1.07 mmol) was added dropwise. The resulting mixture was brought to reflux for 90 min. Then 7.5 ml of solvent was distilled and anhydrous toluene was added (7.5 ml). The solution was again heated under reflux for 1 h and then 7.5 ml of solvent were distilled and anhydrous toluene was added (7.5 ml) and the solution was heated to reflux during 30 min. Then the solution was allowed to cool and the resulting solid was collected by filtration. This solid was thoroughly washed with toluene (100 ml), methylene chloride (100 ml), ethanol (100 ml), and diethyl ether (100 ml), and it was then dried at 50°C under vacuum overnight (phosphorous pentoxide).
Elemental analysis. Found: C, 5.97; H, 1.53; N, 0.42. Equivalent to 0.30 mmol of the linker (referred to nitrogen) per gram of silica gel (linker 0.30 mmol g–1).
3.2.3 Grafting of pybox
To a stirred solution of 3 (126.3 mg, 0.30 mmol) in anhydrous toluene (20 ml) at room temperature, 1 g of freshly prepared aminopropyl-silica (0.30 mmol linker) was added. The resulting mixture was refluxed for 48 h. Subsequently the mixture was cooled at room temperature and filtered. The solid was thoroughly washed with toluene (100 ml), methylene chloride (100 ml) and diethyl ether (100 ml) once each one. Silica gel S1 was then filtered off, and dried under vacuum overnight at 50 °C in the presence of phosphorous pentoxide. Silica S1 was used without further purification.
Elemental analysis. Found: C, 10.07; H, 1.84; N, 1.01.Equivalent to 0.147 mmol of the pybox and 0.134 mmol of free linker (referred to nitrogen) per gram of silica (ligand 0.147 mmol g–1).
3.2.4 Ruthenium complexation
To a stirred solution at r.t. of 300 mg of freshly prepared S1 (ligand 0.049 mmol) in methylene chloride (5 ml), a previously filtered (Acrodisk, PTFE 45 μm) solution of dichloro(p-cymene)ruthenium dimer (15 mg, 0.025 mmol) in methylene chloride (1 ml) was added. The resulting mixture was stirred at room temperature for 24 h more. Silica was then collected by filtration and thoroughly washed with methylene chloride (50 ml) in a Soxhlet apparatus for 24 h. After filtration, S1–Ru was then dried under vacuum overnight at r.t. and phosphorous pentoxide
Ru content analysis: 0.10 mmol g–1 silica.
3.3 S2–Ru
3.3.1 Grafting of the amino spacer
To a stirred suspension of 1.00 g of activated silica gel (Merk 60, Art. No. 109385, 0.040–0.063 mm, heated one night at 140 °C under vacuum) in anhydrous toluene (15 ml) at room temperature, 3-aminopropyltriethoxysilane (0.25 ml, 1.07 mmol) was added dropwise. The resulting mixture was refluxed for 90 min. Then 7.5 ml of solvent was distilled and anhydrous toluene was added (7.5 ml). The solution was again heated under reflux for 1 h and then 7.5 ml of solvent were distilled and anhydrous toluene was added (7.5 ml); the solution was then heated to reflux during 30 min. Then the solution was cooled at 0 °C and the resulting solid was collected by filtration. This solid was thoroughly washed with toluene (100 ml), methylene chloride (100 ml), ethanol (100 ml) and diethyl ether (100 ml) once each one, and then dried at 50 °C under vacuum overnight with phosphorous pentoxide.
Elemental analysis. Found: C, 4.56; H, 1.77; N, 1.47. Equivalent to 1.05 mmol of the linker (referred to nitrogen) per gram of silica gel (linker 1.05 mmol g–1).
3.3.2 Grafting of pybox
To a stirred solution of 3 (170 mg, 0.40 mmol) in anhydrous toluene at room temperature (20 ml), 0.385 g of freshly prepared aminopropyl-silica (0.40 mmol linker) was added. The resulting mixture was refluxed for 48 h. The solution was then cooled at r.t. and the solid was collected by filtration and thoroughly washed with toluene (100 ml), methylene chloride (100 ml) and diethyl ether (100 ml) once each one. Subsequently pybox-silica was dried under vacuum overnight at 50 °C and phosphorous pentoxide.
Elemental analysis (%). Found: C, 19.15; H, 2.64; N, 3.14. Equivalent to 0.469 mmol of pybox and 0.834 mmol of free linker (referred to nitrogen) per gram of silica (ligand 0.469 mmol g–1).
3.3.3 End-capping
To a stirred suspension of 384 mg of freshly prepared pybox-silica (0.46 mmol of ligand) in anhydrous toluene (15 ml) at room temperature, 1,1,1,3,3,3-hexamethyldisilazane (0.27 ml, 1.28 mmol) was added dropwise. The resulting mixture was refluxed for 1 h and then allowed to cool. The resulting silica was collected by filtration and then thoroughly washed with toluene (100 ml), methylene chloride (100 ml), ethanol (25 ml) and diethyl ether (100 ml) once each one. S2 was then filtered off and dried under vacuum overnight at 50 °C and phosphorous pentoxide.
Elemental analysis (%). Found: C, 18.14; H, 2.65; N, 2.90. Equivalent to 0.416 mmol of pybox and 0.822 mmol of free linker (referred to nitrogen) per gram of silica (ligand 0.416 mmol g–1).
3.3.4 Ruthenium complexation
To a stirred solution at r.t. of 200 mg of freshly prepared S2 (ligand 0.082 mmol) in methylene chloride (5 ml), a previously filtered (Acrodisk, PTFE 45 μm) solution of dichloro(p-cymene)ruthenium dimmer (25 mg, 0.041mmol) in methylene chloride (1 ml) was added. The resulting mixture was stirred at room temperature during 24 h. Silica was then collected by filtration and thoroughly washed with methylene chloride (50 ml) in a Soxhlet apparatus during 24 h. S2–Ru was then dried under vacuum overnight at r.t. and phosphorous pentoxide.
Ru content analysis: 0.24 mmol g–1 silica.
4 Conclusions
We have developed a simple, versatile and efficient methodology for the immobilization of chiral pybox systems. This new route opens the way to the preparation of a range of different supported pybox-based catalysts for a wide variety of enantioselective reactions. In the case of silica-supported catalysts, a better control of the pybox dispersion and the number and distribution of free silanols and amino groups is necessary in order to improve the results. In this regard new silica-pybox catalysts with larger surface area and pore size are currently being prepared in our laboratories.
Acknowledgements
This work was made possible by the generous financial support of the C.I.C.Y.T. (Project PPQ2002–04012) and of the Navarra Government (Res.5/2003).