

1 Introduction
Molecular networks in the crystalline phase are infinite periodic molecular architectures resulting from mutual interconnection of molecular tectons [1]. Tectons are active molecular building units containing in their framework energetic and geometric information concerning the designed networks. These molecular assemblies are generated by self-assembly processes taking place between complementary tectons. Molecular tectonics [2] is a branch of supramolecular chemistry dealing with the design of tectons, networks and self-assembly [3] processes leading to the formation of finite or infinite molecular networks. The dimensionality of molecular networks is defined by the number of translations of the connecting nodes, which may be defined as a recognition pattern. Thus 1-, 2- and 3-D networks result from 1, 2 and 3 translations, respectively. The recognition pattern may be classified by the nature of the interaction between consecutive tectons. So far, three classes of networks have been defined. For the first category, called inclusion networks, the recognition pattern is essentially based on van der Waals interactions [4]. The second and third classes based on H- [5] and coordination- [6] bonds are the most frequently investigated.
Let us focus on 1-D coordination networks based on mutual bridging of organic and metallic tectons through the formation of coordination bonds. For these architectures, resulting from a single translation into one direction of space of one or several assembling nodes, in terms of geometry, four possibilities, i.e. linear (a), stair (b) and ‘zigzag’ (c) types and helical (d) may be considered (Fig. 1 ). We have previously demonstrated the possibility of generating a polar crystal based on the linear type arrangement using an enantiomerically pure tecton possessing C2 chirality [7]. In terms of design, a further possibility, which may lead to the formation of a polar crystal, may be based on the use of enantiomerically pure 1-D coordination networks based on a chiral tecton presenting helicity. Pursuing our studies on the design and formation of coordination networks [8] and in particular helical structures [9–11], we have initiated a study dealing with the design of enantiomerically pure coordinating tectons and their assembly into helical structures. Helical assemblies based on coordination processes may either be of the discrete type, such as helicates [12] or infinite networks [13].

Schematic representation of linear (a), stair (b), ‘zigzag’ (c) and helical (d) geometries for 1-D coordination networks.
Here we report the design, synthesis and structural analysis of a series of four tectons 1–4 (Scheme 1) based on isomannide or isosorbide scaffold bearing two pyridine units as well as an enantiomerically pure 1-D helical mercury coordination network.
2 Results and discussion
2.1 Design and synthesis of tectons
Isomannide 5 and isosorbide 6 are two readily available enantiomerically pure diols that may be used as backbones for the design of enantiomerically pure tectons. In particular, the functionalisation of these two scaffolds by two pyridine derivatives as monodentate coordinating units affords coordinating tectons of interest for the generation of coordination networks in the presence of appropriate metal centres. Although in both cases, due to the cis-junction of the two five membered rings, a ‘roof’-type shape is generated for the backbones, they differ by the stereochemistry of the C atoms bearing the OH groups. Indeed, whereas for isomannide 5, the two OH groups point towards the interior of the ‘roof’, for isosorbide 6 they point in opposite directions. Thus, for that reason, the double functionalisation of 5 and 6 by coordination sites leads to two constitutionally identical tectons, but differing by the orientation of the coordinating sites. Enantiomerically pure tectons 1–4 are of the bismonodentate type, based either on the isomannide (tectons 1 and 2) or isosorbide (tectons 3 and 4) backbones. Within each category, the tectons differ by the position of attachment of the pyridine derivative to the backbone. Thus, for tectons 1 and 3, the position 3 of the six-membered ring (with respect to the N atom) is used, whereas for tectons 2 and 4, the pyridine units are connected at position 4. In all cases, the junction between 5 or 6 and the pyridine moiety is ensured by ester groups.
Crown ether type receptors and [2]catenanes based on the isomannide skeleton have been reported [14].
The synthesis of tectons 1–4, rather straightforward, was achieved at room temperature upon treatment of either 5 or 6 by commercially available nicotinoyl chloride 7 or isonicotinoyl chloride 8 as their hydrochloride salts in dry THF and in the presence of Et3N. All four compounds were obtained as crystalline materials. In addition to classical characterisation methods, all four tectons were structurally characterised in the solid state by X-ray diffraction on single crystals (scheme 1) .
2.2 Design and synthesis of coordination networks
For the design of coordination networks that may be defined by (oT, mT), one must consider both the organic tecton oT (shape, number, nature and disposition of coordinating sites, charge) and metallatecton mT (oxidation state, coordination sphere, number and disposition of available coordination sites). The latter may either be a metallic centre often cationic in nature, or a coordination complex possessing available coordination sites. When using a neutral organic tecton oT such as 1–4, for the choice of the metallic tecton mT, two possibilities may be explored: (i) a combination of neutral oT and neutral mT, leading thus to a neutral coordination network of the type (oT, mTL). In that case, the mT is a neutral coordination complex of the type [Mn+Ln–], for which the positive charge on the metal is compensated by the negative charge of the auxiliary ligand L. The other possibility consists in the formation of a polycationic coordination network [(oT, mT)]x+ associated with non-coordinating anions that neither take part in the formation of the network nor are connected to the network by coordination bonds. In that case, the solid would be defined as [[(oT, mT)]x+, xA–] for mono charged anions or as [[(oT, mT)]x+, 0.5 × A2–] for doubly charged anions. Although the latter case has been frequently exploited [6], we have previously demonstrated the use of coordinating anions as a design principle for the generation of neutral coordination networks [7].
As stated above, tectons 1–4 are neutral bismonodentate construction units. In particular, the tecton 1, because of the chiral nature of isomannide, is an enantiomerically pure unit suitable for the formation of helical coordination networks when associated with a metallatecton offering two available coordination sites. For that reason and based on our previous studies [11,15], HgCl2, a neutral metallatecton presenting two available coordination sites, was considered.
2.3 Solid-state structural studies
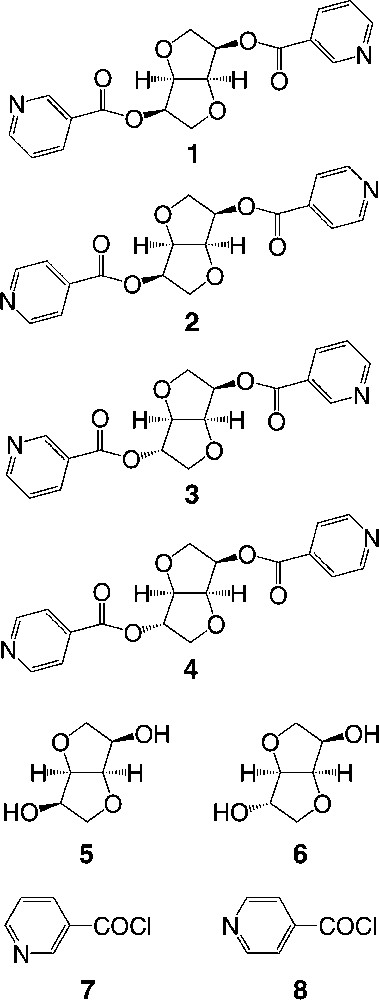
The X-ray diffraction study on single crystal of tectons 1–4 revealed the following features (Table 1). Dealing with tectons 1 and 3 (Fig. 2 ) bearing two nicotinoyl moieties, whereas 1 crystallises in the tetragonal system (space group (P41212), 3 crystallises in the orthorhombic system (P212121). For the other two tectons 2 and 4 (Fig. 3 ) bearing two isonicotinoyl groups, they both crystallise in the orthorhombic system (P212121). For all four cases, the crystal is exclusively composed of the organic tecton and no solvent molecule is present in the lattice. As expected, for tectons 1 and 2 based on the isomannide backbone, the two pyridine units are almost parallel and oriented towards the concave face of the bicyclic unit. For the other two tectons 3 and 4 based on the isosorbide scaffold, the two pyridine groups are oriented in the opposite directions (one towards the interior and the other towards the exterior of the concave skeleton). Except for the orientation of the pyridine units controlled by the backbones and the connecting positions, the other structural characteristics such as bond distances and angles are almost identical for all four tectons 1–4. Indeed, for the ester junctions, dC–O is ranging between 1.34 and 1.35 Å, dC=O varies between 1.19 and 1.20 Å, the OCO angle varies between 123.4 and 124.7°. However, the orientation of the plane of the pyridine ring with respect to plane of the ester varies rather strongly (OCCC dihedral angle of (–10.4°, –10.4°) for 1, (10.3°, –12.2°) for 2, (–5.2°, –18.0°) for 3, (19.6°, 3.2°) for 4).
X-ray data for tectons 1–4 and for the helical coordination network formed between 1 and HgCl2
| 1 | 2 | 3 | 4 | 1–HgCl2 | |
| Chemical Formula | C18H16N2O6 | C18H16N2O6 | C18H16N2O6 | C18H16N2O6 | C18H16N2O6Cl2H |
| Formula weight | 356.33 | 356.33 | 356.33 | 356.33 | 627.83 |
| Crystal system | Tetragonal | Orthorhombic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P41212 | P212121 | P212121 | P212121 | C2 |
| a (Å) | 5.9173(5) | 7.3349(3) | 6.8647(2) | 6.3552(2) | 30.2492(11) |
| b (Å) | 5.9173(3) | 9.5189(4) | 11.0211(4) | 12.0559(4) | 7.4892(6) |
| c (Å) | 44.963(2) | 23.2735(9) | 21.1580(10) | 22.0464(7) | 8.7898(9) |
| α (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 92.365(7) |
| γ (deg) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| U (Å3) | 1574.3(2) | 1624.96(11) | 1600.74(11) | 1689.14(9) | 1989.56(3) |
| Dcalc (g cm–3) | 1.50 | 1.46 | 1.48 | 1.40 | 2.10 |
| Z | 4 | 4 | 4 | 4 | 4 |
| Colour | colourless | colourless | colourless | colourless | colourless |
| Crystal dimensions (mm) | 0.34 × 0.28 × 0.18 | 0.36 × 0.24 × 0.17 | 0.20 × 0.20 × 0.12 | 0.26 × 0.22 × 0.12 | 0.34 × 0.16 × 0.16 |
| μ(Mo Kα) (mm–1) | 0.12 | 0.11 | 0.11 | 0.11 | 8.04 |
| Temperature (K) | 173.00 | 173.00 | 173.00 | 173.00 | 173.00 |
| Number of data measured | 2343.00 | 4732.00 | 4646.00 | 4884.00 | 7004.00 |
| Number of data with I > 2 σ(I) | 1687.00 | 3728.00 | 3804.00 | 3807.00 | 5407.00 |
| R | 0.040 | 0.044 | 0.043 | 0.042 | 0.039 |
| Rw | 0.080 | 0.11 | 0.096 | 0.091 | 0.115 |
| GOF | 1.013 | 1.089 | 1.076 | 1.013 | 1.093 |
| CCDC No. | 216 738 | 216 737 | 216 739 | 216 740 | 216 741 |

Crystal structures of tectons 1 (top) and 3 (bottom). H atoms are omitted for clarity. For bond distances and angles, see text.
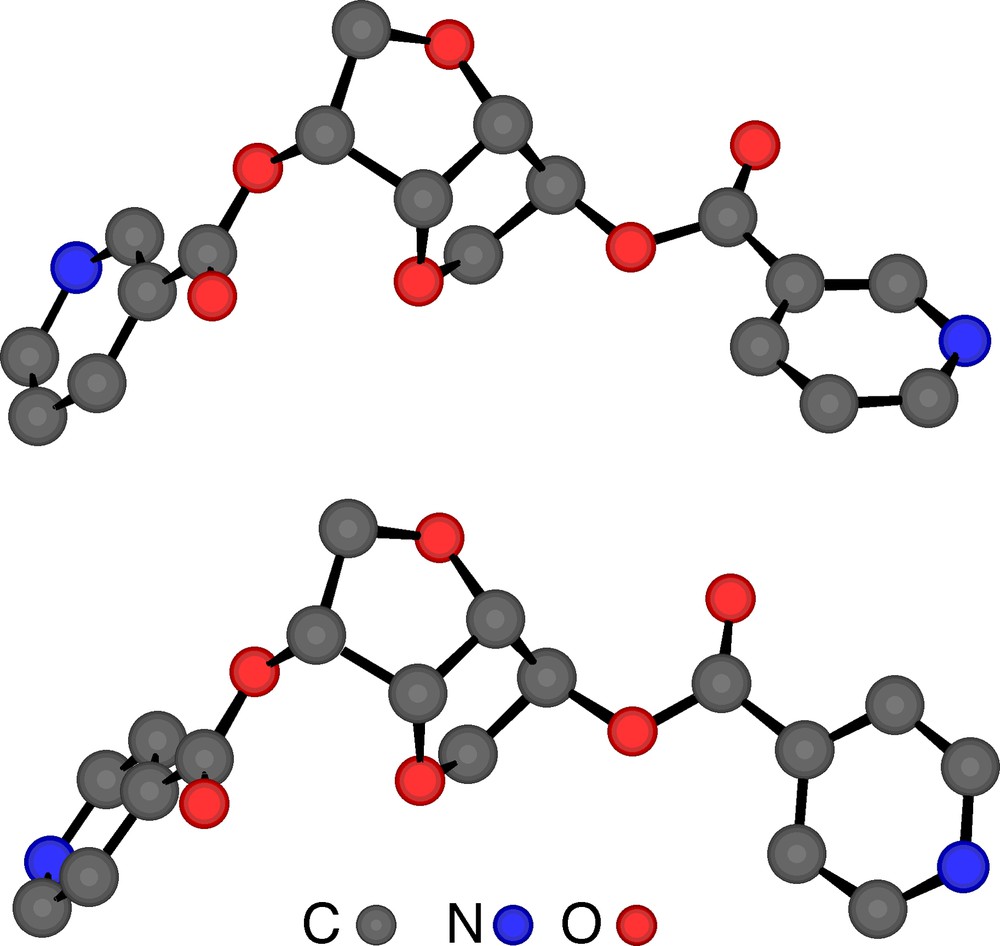
Crystal structures of tectons 2 (top) and 4 (bottom). H atoms are omitted for clarity. For bond distances and angles, see text.
Upon diffusion at room temperature of a solution of the tecton 1 in CHCl3 into a solution of HgCl2 in EtOH, colourless crystals were obtained and studied by X-ray diffraction on single crystal. The crystal is exclusively composed of 1 and HgCl2 and no solvent molecule is present in the lattice (Table 1). As expected, the mutual interconnection between the organic tecton 1 and HgCl2 metallatecton leads to an enantiomerically pure neutral single stranded infinite helix with P helicity (Fig. 4 ). The observed P helicity results from the defined stereochemistry of the isomannide unit and consequently of the tecton 1. The infinite helical network crystallises in the non-centrosymmetric polar space group C2 in the monoclinic system. For the tecton 1, the two pyridine units connected to the isomannide backbone through ester junctions (average dC–O of ca 1.33 Å and average dC=O of ca 1.20 Å, OCO angle of 124.2° and 125.2°) are almost parallel and convergently oriented towards the concave face of the backbone. The pyridine rings are tilted with respect to plane of the ester (OCCC dihedral angle of 0.0° and –9.3°. The infinite 1-D coordination network with helical geometry may either be regarded as resulting from bridging of the organic tectons 1 by HgCl2 units or conversely as interconnection of HgCl2 complexes by the organic tecton 1. The coordination sphere around both crystallographically non-equivalent Hg2+ cations is composed of two N atoms belonging to two consecutive organic tectons 1 (dHg–N varying between ca 2.37 Å and ca 2.43 Å) and two chloride anions (average dHg–Cl of ca 2.37 Å). Both crystallographically non-equivalent Hg2+ cation adopts a distorted tetrahedral (Cl–Hg–N angle varying between 96.6° and 97.9°, Cl–Hg–Cl angle varying between 148.0° and 150.2° and N–Hg–N angle varying between 121.2° and 121.6°). The distance between two consecutive Hg2+ cations is 5.76 Å. The helix extends along the c axis. The fragment that determines the pitch (8.79 Å) of the helix is composed of two HgCl2 and two tectons 1.
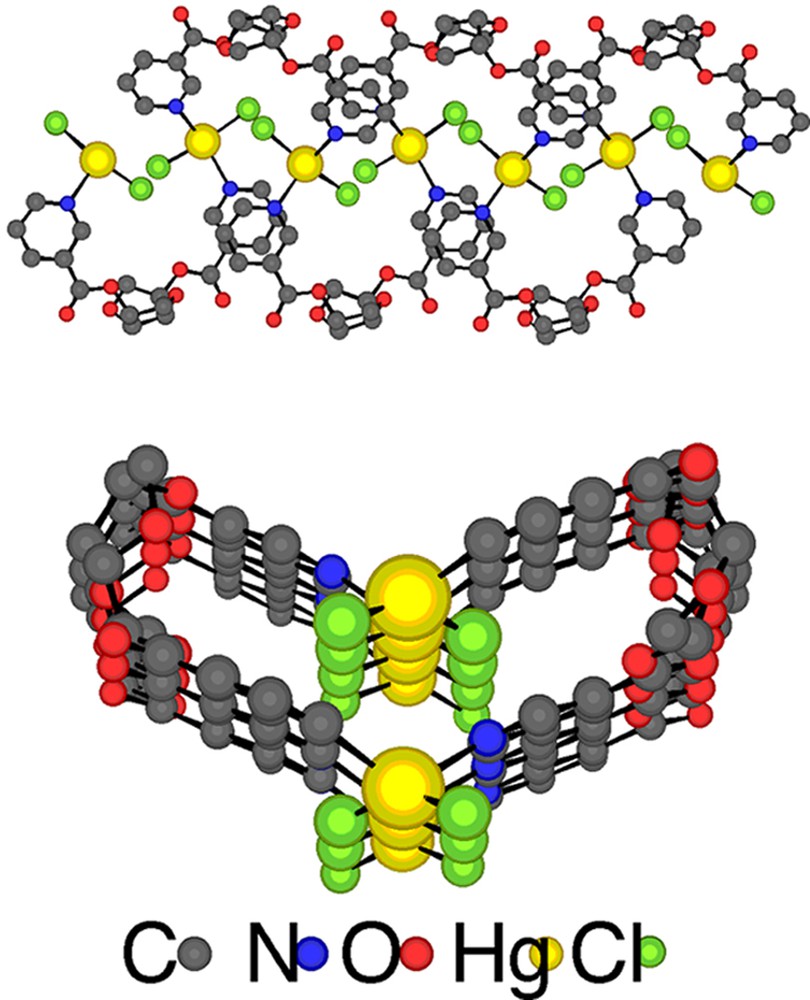
Perpendicular (top) and along the helix axis (bottom) views of a portion of the enantiomerically pure infinite helical 1-D coordination network (P helicity) formed upon bridging of consecutive tectons 1 by HgCl2. H atoms are omitted for clarity. For bond distances and angles, see text.
Let us consider the parallel packing of infinite helical strands (Fig. 5 ). In principle, one may envisage several possibilities. For a racemate (presence of the same amounts of helices with P and M helicity: (P,M)), the syn-parallel arrangement will lead to a polar assembly (Fig. 5a), whereas the anti-parallel packing will generate a non-polar architecture (Fig. 5b). The prefix syn or anti is used in order to define the orientation of the polar axis of the helix. For the homochiral situation (exclusive presence of enantiomerically pure helices either with P or M helicity), again depending on the orientation of the polar axis of the helices, either polar (Fig. 5c and d) or apolar assemblies (Fig. 5e and f) may be formed. In the case of the enantiomerically pure helical arrangement obtained upon self-assembly of the tecton 1 and HgCl2, the packing of the helical strands takes place in the syn-parallel mode (of the type c, Fig. 5) and, consequently, the crystal formed is polar (Fig. 6 ). The analysis of packing revealed no specific interaction between helical strands in the crystal.
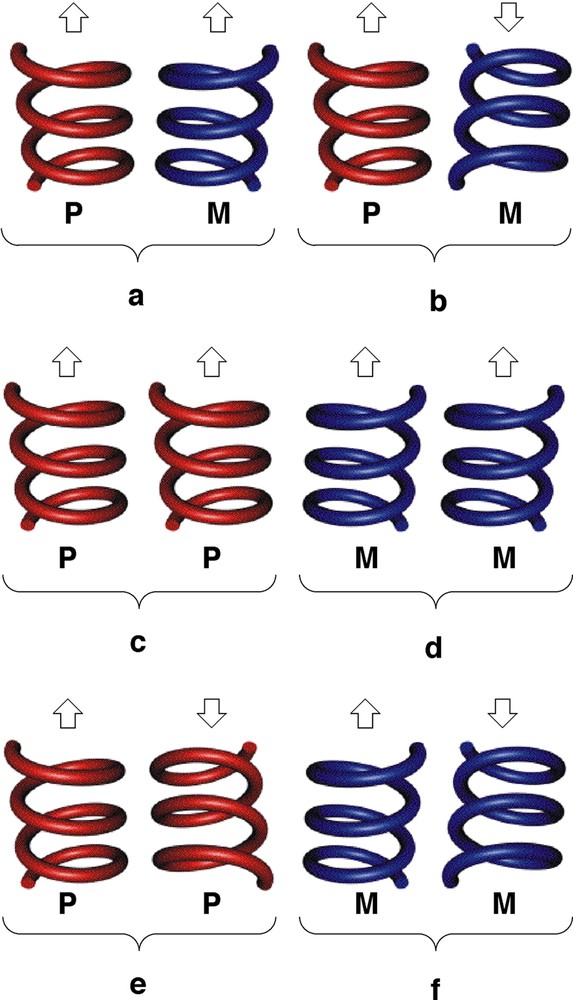
Schematic representation of different possibilities of arrangement of infinite helical strands. Polar (a) and apolar (b) arrangements for a racemate (heterochiral mixture), polar (c and d) and apolar (e and f) for a homochiral system. The arrow represents the direction of polarity.
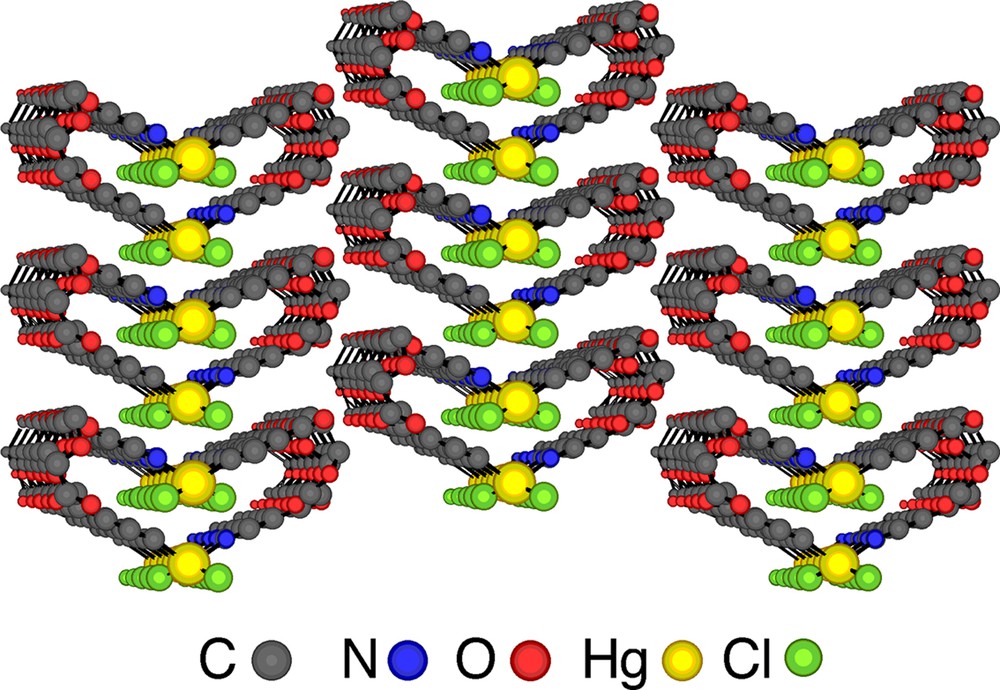
A parallel view on the lateral packing of enantiomerically pure single stranded helices showing the syn-parallel arrangement of helical strands and the formation of a polar crystal. H atoms are omitted for clarity.
The combination of 1 with HgCl2 is rather different from the combination of 2 and HgCl2. Indeed, using HgCl2 as a metallatecton, whereas the tecton 1 leads to the formation of a single stranded helical architecture, the tecton 2 generates a triple helix [11].
2.4 Conclusion
The synthesis of four new pyridine containing tectons 1–4 based on isomannide (1,2) and isosorbide (3,4) skeletons was achieved and their structure elucidated in the solid state by X-ray diffraction on single crystals. Under self-assembly conditions and in the presence of HgCl2, the tecton 1 based on the isomannide backbone bearing two nicotinoyl moieties generates in the solid state an enantiomerically pure 1-D coordination network of the helical type. The syn-parallel packing of the single stranded helices leads to the formation of a polar crystal. The formation of either chiral metallamacrocycles or infinite coordination networks using other metals is currently under investigation.
3 Experimental section
3.1 Synthesis of tectons
3.1.1 General
All commercially available reagents were purchased and used without further purification. 1H and 13C NMR spectra were recorded on Bruker spectrometers at 400 and 300 MHz and at 50 MHz, respectively. Microanalyses were performed by the Service de microanalyses de la fédération de recherche chimie, ‘université Louis-Pasteur’, Strasbourg, France.
Polarimetric measurements were performed on a JASCO DIP370 digital polarimeter. The optical rotation was measured at 20 °C at the concentration of 1.00 mol l–1 in CHCl3.
3.1.2 General procedure for the synthesis of compounds 1–4
To a solution (150 ml) of 5 or 6 (1 g, 6.8 mmol) in dry THF, 7 or 8 (3.05 g, 17.1 mmol, 2.5 equiv) was added as its hydrochloride salt and the mixture was stirred at room temperature for 15 min. To the mixture Et3N (4 ml) was added and stirring was further continued overnight. After evaporation to dryness, to the residue was added a saturated aqueous solution of Na2CO3 (40 ml) and the mixture extracted with CH2Cl2 (3 × 30 ml). The evaporation to dryness left a residue that was suspended in CH2Cl2. The pure compounds 1–4 were obtained by filtration over alumina as white solids which were further recrystallised by slow evaporation of CHCl3/hexane 3:2 mixture containing the desired compound.
1 (1.46 g, 60% yield): mp 130 °C, [α]D = +223.3; anal. calc. for C6H16N2O6.(mw = 356.33): C 60.67%; H 4.53%, N 7.86%, found: C 59.95%; H 4.60%, N 7.99%; 1H–NMR (25 °C, CDCl3): δ (ppm) 4.01 (m, 2H), 4.14 (m, 2H), 4.89 (m, 2H), 5.34 (m, 2H), 7.40 (m, 2H), 8.32 (m, 2H), 8.79 (m, 2H), 9.26 (dd, 2H, J = 0.8 Hz and 2.2 Hz); 13C-NMR (25 °C, CDCl3) δ (ppm): 70.6, 74.4, 80.6, 123.3, 125.3, 137.2, 151.0, 153.8, 164.6.
2 (1.5 g, 62% yield): mp 145 °C, [α]D = +198.6; anal. calc. for C6H16N2O6 (mw = 356.33): C 60.67%; H 4.53%, N 7.86%, found: C 59.82%; H 4.61%, N 7.82%; 1H-NMR (25 °C, CDCl3): δ (ppm) 4.00 (m, 2H); 4.13 (m, 2H), 4.90 (m, 2H), 5.35 (m, 2H), 7.87 (dd, 4H, J = 1.6 and 4.4 Hz), 8.79 (dd, 4H, J = 1.6 and 4.4 Hz); 13C–NMR (25 °C, CDCl3) δ (ppm): 70.5, 74.7, 80.5, 122.8, 136.5, 150.7, 164.5.
3 (1.41 g, 58% yield): mp 129 °C, [α]D = +32.0; anal calc. for C6H16N2O6 (mw = 356.33): C 60.67%; H 4.53%, N 7.86%, found: C 59.82%; H 4.61%, N 7.82%; 1H-NMR (25 °C, CDCl3): δ (ppm) 4.08 (m, 4H), 4.67 (d, 1H, J = 4.75 Hz), 5.08 (t, 1H, J = 5.1 Hz), 5.41 (m, 2H), 8.29 (m, 2H), 9.20 (d, 1H, J = 0.8 Hz), 9.25 (d, 1H, J = 2.2 Hz); 13C–NMR (25 °C, CDCl3) δ (ppm): 70.5, 74.5, 80.6, 123.3, 125.3, 137.2, 151.0, 153.8, 164.6.
4 (1.29 g, 53% yield): mp 138 °C, [α]D = +41.8; anal calc. for C6H16N2O6 (mw = 356.33): C 60.67%; H 4.53%, N 7.86%, found: C 60.40%; H 4.46%, N 7.91%; 1H-NMR (25 °C, CDCl3): δ (ppm) 4.07 (m, 4H), 4.67 (d, 1H, J = 4.7 Hz), 5.06 (t, 1H, J = 5.1 Hz), 5.43 (q, 1H, J = 5.2 Hz ), 5.49 (s, 1H), 7.80 (d, 2H, J = 4.7 Hz), 7.86 (d, 2H, J = 4.7 Hz), 8.78 (m, 4H); 13C-NMR (25 °C, CDCl3) δ (ppm): 70.7, 73.2, 75.1, 79.0, 81.0, 86.0, 122.8, 136.5, 150.7, 164.3.
3.2 Generation of networks
At room temperature, upon slow liquid/liquid diffusion of a solution of HgCl2 (2 mg) in EtOH (2 ml) into a solution of the tecton 1 (5 mg) in chloroform (1 ml), colourless single crystals were obtained after several days.
3.3 X-ray structure analysis
X-ray diffraction data collection was performed at 173 K on a Kappa CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo Kα radiation (see Table 1). All structures were solved by direct methods using the Nonius Maxus Package 4.3. All non-H atoms were refined anisotropically. Atomic coordinates, bond lengths and angles, and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre (see below).
Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif files No. CCDC 216737-216741, and can be obtained by contacting the CCDC.
Acknowledgements
We would like to thank M. Henry for helpful discussions and the ‘Université Louis-Pasteur’ as well as the French ‘Ministère de la Recherche et de la Technologie’ for financial support.