

1 Introduction
Random inorganic networks consisting of covalently bonded non-metals, such as silicon, boron, nitrogen and carbon, represent a rather young class of high-performance materials with outstanding thermal, chemical and mechanical properties [1,2]. Due to their low density and durability at elevated temperatures, those ceramics suggest themselves for applications under extreme conditions like in heat engines, in particular turbines. However, multinary solids in the system Si/B/N/C cannot be produced from the pure elements or the respective binary compounds, since the latter melt incongruently, and, at lower temperatures, exhibit small self-diffusion coefficients [3,4]. So far, the only approach known to the synthesis of homogeneous multinary non-oxide ceramics is the polymer route [5–7], which comprises the polymerization of one or more metalorganic monomers and subsequent pyrolysis of the resulting preceramic polymer. Following this path, Winter et al. obtained the first Si/N/C fibers by means of spinning and pyrolyzing inorganic polymers, obtained from the aminolysis or ammonolysis of methylchlorosilanes [8]. Similarly, Yajima et al. converted spinnable polycarbosilanes into (carbon-rich) SiC fibers, which were the first commercially available ceramic fibers (Nicalon™) [9]. In addition, preceramic polymers have been successfully processed to coatings [10] or can be employed for the fabrication of microstructures using soft lithographic techniques [11].
The performance of multinary ceramics like the quaternary Si/B/N/C random networks depends strongly on how the material is synthesized. If the preparation of a preceramic polymer proceeds via co-condensation of molecular precursors containing only one electropositive element (silicon or boron), spatial inhomogeneities often are encountered due to different velocities of substitution reactions of the individual molecular species. Such inhomogeneities usually induce phase separation and decomposition of the multinary ceramic into its border phases during pyrolysis or thermal load resulting in failure of the final ceramic workpiece. This problem can be overcome by using the so-called single-source precursors that, after polymerization with a cross-linking reagent, provide preceramic polymers with a perfect homogeneous distribution of all constituting elements on an atomic scale. These special monomers already contain silicon and boron in the ratio desired for the final ceramic, linked together via a bridging atom (mostly carbon or nitrogen). As both cations are fixed to the bridging group by strong covalent bonds, the backbone of the precursor survives the whole sequence of processing (polymerization and pyrolysis), thus leading to a highly homogeneous distribution of all elements in the final ceramic.
In the last 10 years a large number of new single-source precursors for Si/B/N/C ceramics with different Si/B ratios have been developed using various synthetic routes [12]. Cross linking of the organometallic compounds, usually with methylamine, is yielding polyborosilazanes or polyborocarbosilazanes, depending on the bridging atom between silicon and boron. As a special feature, pyrolysis of these polymers at 1400 or 1500 °C in an inert atmosphere provides amorphous ceramics that, although being unstable with respect to transformation or decomposition into crystalline phases, show a distinct kinetic stability even at elevated temperatures. The amorphous state as well as the high resistivity towards crystallization is clearly a result of the strong covalent bonds formed by the elements that constitute the random network. Hence, the total enthalpy of formation lies predominantly in localized bonds so that the energetic benefit from long-range order is negligibly small, thus minimizing the thermodynamic driving force towards crystallization. As a consequence, amorphous Si/B/N/C materials exhibit both, a reduced brittleness (due to the lack of lattice planes guiding crack propagation) [2] and an excellent creep resistance [1], since no grain boundaries are present, which allows for gliding of crystallites.
The incorporation of carbon into an Si/B/N random network plays a crucial role for the thermal durability of the resulting carbonitridic ceramic, as can be seen from the thermal stabilities of Si3B3N7 [13] compared to SiBN3C [14]. While the ternary nitride, as obtained via polycondensation of the single-source precursor Cl3Si–NH–BCl2 (TADB [13]) and ammonia, starts to decompose at 1650 °C, the quaternary material, synthesized from TADB and methylamine is stable up to 1750 °C, with respect to weight loss. Further incorporation of carbon by polymerizing the carbon-rich precursor Cl3Si–CH(CH3)–BCl2 (TSDE [15]) with methylamine results in a ceramic, with the empirical formula Si3B3N7C6, whose thermal stability again is increased by approximately 100 K compared to SiBN3C. According to this, it seems likely that compounds with a higher carbon content exhibit an improved thermal durability.
Generally, the carbon content in Si/B/N/C ceramics can be controlled by both the constitution of the precursor molecule and the cross-linking reagent. However, the amount of carbon cannot be increased simply by choosing longer alkyl chains, as terminal alkyl groups usually get lost during pyrolysis and, in addition, precipitation of graphite may occur. In order to achieve an efficient incorporation of carbon into the random network during polymerization and pyrolysis, the single-source precursors should contain carbon as a bridging methylene group so that each carbon atom is connected to the constituting network via two covalent bonds instead of only one bond, which is the case when carbon is introduced through a methyl group. In this contribution, we present the synthesis of new single-source precursors with a methylene group between silicon and boron for the preparation of carbon-rich amorphous Si/B/N/C ceramics with an improved stability at high temperatures.
2 Results and discussion
2.1 Syntheses of the single-source precursors Cl3Si–CH2–BCl2 and (CH3)Cl2Si–CH2–BCl2
2.1.1 Designing the synthesis
Two molecules of simple constitution, but meeting the preconditions discussed above are shown in Fig. 1. Both single-source precursors contain boron and silicon in the ratio of 1:1, interconnected via a bridging methylene group. To enable cross linking of the precursors with methylamine, the remaining bonding sites of silicon and boron are saturated with chlorine atoms. Substitution of a methyl group for a chlorine atom in Cl3Si–CH2–BCl2 (trichlorosilyl dichloroboryl methane, TSDM) leads to (CH3)Cl2Si–CH2–BCl2 (dichlorosilyl dichloroboryl methane, DSDM), which, after conversion into the final ceramic, provides a material with increased carbon content compared to that one derived from TSDM. Moreover, the lower number of chlorine functions in DSDM available for polycondensation offers an opportunity for tuning the rheological properties of the polymers.

Simple acyclic single-source precursors with a methylene bridge and (a) five as well as (b) four chlorine functions as cross-linking sites.
For the selective syntheses of the precursors TSDM and DSDM, metathesis reactions between the respective organometallic silanes and boron halide are virtually the only feasible way (Fig. 2a). Although in principle a metathesis reaction between an organometallic borane and a silicon halide is also possible (Fig. 2b), in the former route less preparative difficulties are involved, because of the better availability and also the higher stability of a metallated alkyl chlorosilane.
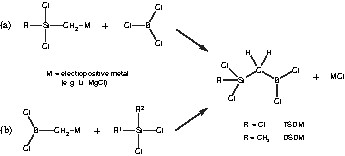
Synthetic routes for the preparation of the single-source precursors TSDM and DSDM through metathesis reactions between (a) an organometallic silane and a boron halide or (b) an organometallic borane and a silicon halide.
Nevertheless, the synthesis of a metalorganic trichlorosilane Cl3Si–CH2–M is highly challenging, because of possible (self-) polymerization. Thus, the preparation of the Grignard compound Cl3Si–CH2–MgCl starting from chloromethyl trichlorosilane with magnesium directly leads to a highly cross-linked chloropolycarbosilane [16]. In the same way, reaction of Cl2(CH3)Si–CH2Cl and Cl(CH3)2Si–CH2Cl with magnesium respectively, ends in viscous polymers as well as in small amounts of cyclic carbosilanes in different sizes [17,18].
Up to now, there is no synthetic approach known in the literature avoiding the detrimental self-condensation of such bifunctional compounds.
2.1.2 Preparation of the Grignard compound
In order to suppress self-condensation of the metallated alkylchlorosilane at the best possible rate, one has to take good care of the reaction conditions. Firstly, the M–C bond of the metalorganic chlorosilane should not be too polar, so that the reactivity towards the chlorine function in the same molecule is somewhat decreased. Thus, Grignard reagents are particularly well suited; moreover, they are also easy to prepare. Secondly, the reaction temperature has to be chosen as low as possible, but high enough to ensure reaction of the silylalkyl halide with magnesium. Analogously, the concentration of the Grignard reagent has to be kept as low as possible (dilution principle), but at the same time high enough to enable the addition of the silylalkyl halide to magnesium. Finally, the competing undesired polycondensation of the Grignard compound can be suppressed if the oxidative addition step is accelerated. This can be achieved by using magnesium powder rather than magnesium turnings, because the powder reveals a higher active surface.
2.1.3 Synthesis of the target molecules
Unfortunately, the direct reaction of the Grignard reagent Cl3Si–CH2–MgCl or Cl2(CH3)Si–CH2–MgCl with trichloroborane faces some obstacles. While the preparation of the magnesium organyl has to be carried out in an ether solvent (e.g., diethyl ether or THF) trichloroborane, as a strong Lewis acid, reacts with ethers, even at low temperatures [19]. This contrariness can be resolved by exchanging trichloroborane for a chloroborane with reduced Lewis acidity, e.g. a dialkoxychloroborane. The organoborane dialkylester obtained this way is then halogenated to yield the desired single-source precursors TSDM and DSDM, respectively. The corresponding reaction steps are summarized in Fig. 3a and b. It should be noticed that the chlorination step leads to an equilibrium, from which the product cannot be distilled off since trichloroborane has the lowest boiling point (b.p.). In order to shift the equilibrium reaction quantitatively towards the products, catalytic amounts of aluminum trichloride must be added, causing the present alkoxydichloroborane to decompose completely into boron trioxide, trichloroborane and an alkyl chloride [20,21] (Fig. 3c).
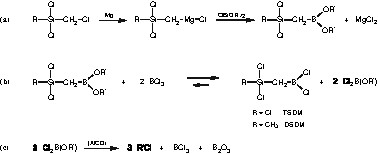
Reaction steps involved for the preparation of the single-source precursors TSDM and DSDM.
The length of the alkyl rest R′ in the alkoxy group determines both the stability of the alkoxydichloroborane [20] and the b.p. of the intermediate organoborane dialkylester. The ease of decomposition of the alkoxydichloroborane increases with the chain length of R′ and also in the order tert. > sec. > prim. Choosing R′ = ethyl is ensuring an optimal combination of rapid decomposition of the alkoxydichloroborane and formation of a distillable organoborane dialkylester.
2.2 Cross linking of the single-source precursors
The role of carbon for the high-temperature stability of silicon boron carbonitrides has already been discussed in the introduction. In order to further increase the carbon content starting from a given precursor molecule, TSDM and DSDM are polymerized with methylamine, since this amine is the most simple carbon-containing cross-linking agent. Due to the dehydrohalogenation reactions involved, HCl forms and adds to excessive methylamine, leading to the precipitation of methylammonium hydrochloride. The resulting polyborocarbosilazane is soluble in organic solvents, e.g. hexane, thus facilitating the separation of the solid methylammonium salt by filtration. After removal of the organic solvent, the polymer usually remains as a colorless viscous liquid that can be solidified by further thermal treatment.
2.2.1 Spectroscopic characterization of the polyborocarbosilazanes
The infrared (IR) spectra of the solid polymers derived from TSDM and DSDM are shown in Table 1. Due to small local structural variations, the absorption of each functional group in the polymer is somewhat broadened compared to the appropriate band present in the IR spectra of the pristine precursor. The spectra of the polymers are dominated by the symmetric and antisymmetric stretching modes of the methyl groups introduced via methylamine as well as by the Si–N and B–N stretching modes. The absorptions at approximately 3440 cm–1 are related to terminal NH groups and their intensities can be utilized to estimate the degree of cross linking inside the polymer. The absorptions at approximately 1100 cm–1 clearly indicate the presence of B–C bonds, which gives evidence for the structural feature Si–CH2–B having survived the whole process of the aminolysis and thermal treatment. However, the mandatory stretching modes of the Si–C bonds cannot be resolved due to their similarity to Si–N modes.
Most important IR absorptions of the polymers after cross linking of TSDM and DSDM with methylamine (wavenumbers in (cm–1); vs: very strong; s: strong; m: medium; w: weak, vw: very weak)
| Vibration | TSDM polymer | DSDM polymer |
| ν (NH) | 3433 m | 3448 vw |
| νas (CH3) | 2962 s | 2925 vs |
| νas (CH3) | 2889 s | 2855 s |
| νs (CH3) | 2808 m | 2813 w |
| δas (CH3) | 1486 m | 1464 m |
| ν (BN) | 1351 m | 1377 m |
| δs (CH3) | 1262 s | 1253 m |
| ν (BC) | 1097 s | 1099 m |
| ν (SiC)/ν (SiN) | 803 s | 789 m |
In order to get a more significant proof for the methylene bridge between silicon and boron both, 29Si and 11B MAS NMR spectra of the TSDM-derived polymer have been recorded (Fig. 4). While the 29Si MAS spectrum exhibits a single broad resonance centered at –15.1 ppm, clearly indicating that silicon is tetrahedrally coordinated by one carbon and three nitrogen atoms [22], the 11B MAS spectrum is characterized by a powder pattern indicative of second-order quadrupolar interaction. Simulation of the lineshape (dashed line in Fig. 4) results in a quadrupolar coupling constant of 2.88 MHz, an asymmetry parameter of 0.23 and an isotropic chemical shift value of 33.0 ppm, confirming that boron is trigonally planar coordinated by one carbon and two nitrogen atoms. These findings suggest that the structural feature Si–CH2–B is still present in the TSDM polymer and should also be present in the DSDM polymer, since DSDM has been polymerized exactly under the same conditions.
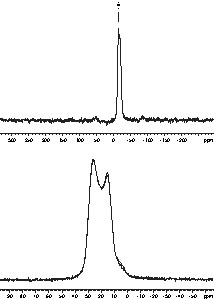
29Si (top) and 11B MAS NMR spectra of the polymer as derived from TSDM. Simulation of the 11B spectrum (dashed line) results in δiso = 33.0 ppm, CQ = 2.88 MHz, ηQ = 0.23.
2.2.2 Thermal degradation of the polyborocarbosilazanes
The pyrolytic conversion of the polymers into the desired amorphous ceramics was monitored by differential thermal analysis combined with thermogravimetric analysis and mass spectrometry (DTA/TG/MS). According to these measurements, the thermal degradation of both, the TSDM and the DSDM polymer proceeds in two well-resolved stages (Fig. 5), with an overall ceramic yield of 70.3% and 47.5%, respectively. From 200 to 450 °C, the polycondensation, accompanied by methylamine elimination, is completed, resulting in mass losses of 7.1% and 18.1% for the TSDM and the DSDM polymer, respectively. At about 600 °C, the highest weight loss occurs due to the evolution of methane from terminal methyl groups. Since the number of methyl groups in the DSDM polymer is higher than in the TSDM polymer, the weight loss during this step of degradation accounts for 34.4% and 22.6%, respectively. The degradation of the polymers in two steps is in good accordance with earlier investigations on the conversion of polyborosilazanes to silicon boron carbonitrides [23]. However, a therein reported third step with a mass loss of about 4% between 1100 and 1300 °C due to the emission of nitrogen, does not occur during the degradation of the polymers derived from TSDM and DSDM. This behavior goes back to the structure of precursor molecules from which the different polyborosilazanes have been synthesized. Due to the carbon bridge in TSDM and DSDM, the corresponding polymers exhibit a relatively low nitrogen content compared to the formerly investigated polymers. As a consequence, all nitrogen present in the polymer is incorporated into the final random network.
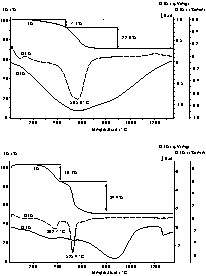
DTA/TG curves of the TSDM (top), and of the DSDM polymer.
2.3 Carbon-rich Si/B/N/C ceramics
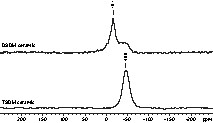
Heat treatment of the TSDM and DSDM polymer up to 1500 °C converts the preceramic polymers into black Si/B/N/C ceramics. As can be seen from Fig. 6, both materials are X-ray amorphous after the pyrolysis and calcination procedure. The surface of the ceramic grains is irregular with sharp edges and no noticeable porosity (Fig. 7).
XRD powder patterns of the prepared Si/B/N/C ceramics after annealing at 1500 °C.

Scanning electron micrographs of the ceramics derived from TSDM (top) and DSDM.
The composition of the newly synthesized ceramics is shown in Table 2. As expected, the 1:1 ratio Si/B remains the same in the final ceramic as predetermined in the single-source precursors TSDM and DSDM. Furthermore, carbon is incorporated into the random network to a high degree, leading to two new carbon-rich silicon boron carbonitrides of idealized composition Si3B3N7C5 and Si3B3N5C7. The high efficiency of the incorporation of carbon can be ascribed to the methylene bridge present in the precursor molecules. Due to the linkage to two neighboring atoms (silicon and boron) via two strong covalent bonds, carbon becomes an inherent part of the preceramic polymer, and also of the final ceramic. Using DSDM, additional carbon has been introduced through a terminal methyl group, and the carbon content of the resulting ceramic is noticeably increased from 20.8 to 30.8 mass-%.
Composition of the ceramics derived from TSDM and DSDM (indicated as mass-%)
| Ceramic derived from | Si | B | N | C | O | ∑ | Empirical formula | |
| Exactly | Approximately | |||||||
| TSDM | 30.7 | 11.6 | 34.8 | 20.8 | 1.4 | 99.3 | Si1.0B1.0N2.3C1.6O<0.1 | Si3B3N7C5 |
| DSDM | 30.5 | 12.3 | 26.8 | 30.8 | 0.8 | 101.2 | Si1.0B1.0N1.7C2.3O<0.1 | Si3B3N5C7 |
The macroscopic densities of the ceramics (standard deviation in parentheses), measured using a helium pyknometer, are rather similar (1.9272(4) g cm–3 for the TSDM-derived ceramic, 1.9306(2) g m–3 for the DSDM ceramic). Remarkably, these values are approximately 30% below the densities of comparable crystalline phases like SiC or Si3N4, although no macroscopic porosity in the silicon boron carbonitrides can be found.
The high-temperature behavior of the ceramics was tested with a DTA/TG thermal analysis in a helium flow (Fig. 8). On heating the samples at a rate of 10 K min–1) up to 2000 °C, a final weight loss of 19% and 12% for the TSDM and the DSDM ceramic (pretreated at 1500 °C), respectively, occurs indicating a significant increase in temperature stability as compared to pre-described Si/B/N/C materials [13,24,25]. In line with our conceptual design, a further increase of the carbon content in the random Si/B/N/C network leads to an improved high-temperature stability. In neither of the DTA curves, peaks that might be attributed to endothermic or exothermic effects were observed. X-ray diffraction (XRD) analysis of the TSDM ceramic after temperature treatment at 2000 °C indicates the formation of Si3N4 and SiC, whereas in the DSDM ceramic only SiC is detectable. The presence of Si3N4 beyond its decomposition temperature (1600 °C at ambient pressure) has already been observed in the system Si/B/N/C [26] and was ascribed to the encapsulation of Si3N4 crystallites, leading to an increase of the internal nitrogen pressure and consequently to an increase of the decomposition temperature.
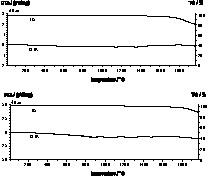
DTA/TG curves up to 2000 °C for the TSDM (upper) and the DSDM ceramic (measured in a helium flow at 10 K min–1).
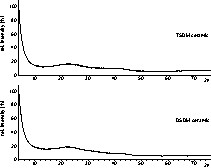
In order to check the oxidation behavior, the two silicon boron carbonitrides were heated at a rate of 10 K min–1 up to 1300 °C in a flow of pure oxygen (Fig. 9). The TSDM ceramic showed no effect that might indicate an oxidation process or decomposition. A small increase in weight of 2.4% at 1300 °C can be attributed to the formation of a passivating surface layer rich in oxygen and silicon [2], preventing the ceramic from being further oxidized. For the carbon-richer DSDM ceramic, the increase in weight of 2.8% at 1300 °C is also due to the formation of a thin passivating layer. Moreover, at 700 °C a slight decrease in weight of 6.8% is noticed, suggesting an oxidation process during which a volatile component is evolved.

Oxidation behavior of the TSDM (upper) and the DSDM ceramic up to 1300 °C (measured in an oxygen flow at 10 K min–1).
In order to gain structural information about the short-range order, the random networks were investigated by means of solid-state MAS NMR experiments on 29Si and 11B [27,28]. The 29Si MAS spectra (Fig. 10) of both, the TSDM and the DSDM ceramic show the typical inhomogeneous linebroadening of amorphous compounds, due to local deviations of bond lengths and bond angles around silicon. The 29Si MAS spectrum of the TSDM ceramic exhibits a single resonance at –46.5 ppm, indicative for silicon, which is tetrahedrally coordinated by four nitrogen atoms [22]. In contrast to this, the 29Si MAS spectrum of the DSDM ceramic shows one intense signal at –16.1 ppm, which can be ascribed to a tetrahedral SiNC3 coordination sphere, as well as a broad resonance covering the whole area of chemical shifts for mixed tetrahedral C/N coordination spheres SiN3C, SiN2C2, SiNC3 and also SiN4.
Single pulse 29Si MAS spectra of the ceramics derived from TSDM and DSDM.
The 11B MAS spectrum of the TSDM ceramic shows a powder pattern indicative of a second-order quadrupolar interaction (Fig. 11). Simulation of the lineshape results in a quadrupolar coupling constant of 2.88 MHz, an asymmetry parameter of 0.16 and an isotropic chemical shift value of 28.7 ppm. In accordance with the 11B MAS NMR spectra of known Si/B/N/C ceramics [29,30] and h-BN, the data confirms that boron is trigonally planar coordinated by three nitrogen atoms. The DSDM ceramic represents a remarkable exception, since its 11B spectrum cannot be simulated with only one resonance but rather with two signals (Fig. 12). As a result, beside a small amount of tetrahedrally coordinated boron atoms, the majority of boron atoms is coordinated trigonally planar. Although the isotropic chemical shift value of 29.9 ppm is somewhat low for a BN2C coordination sphere, this value may be interpreted together with a significant asymmetry factor of 0.26 in terms of a mixed C/N coordination of boron. Likewise, the chemical shift value of –0.5 ppm for the tetrahedrally coordinated boron atoms is in good agreement with the 11B chemical shift value of –0.1 for a pyrazolyl borate with boron in a tetrahedral BN3C coordination sphere [31].
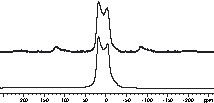
11B MAS spin echo spectrum of the TSDM ceramic. The simulation of the spectrum (lower line) results δiso = 28.7 ppm, CQ = 2.88 MHz, ηQ = 0.16.
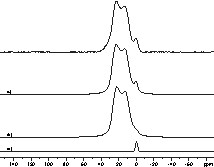
11B MAS spin echo spectrum of the DSDM ceramic. The simulation (c) of the spectrum results in the components (a) δiso = –0.5 ppm, and (b) δiso = 2.99 ppm, CQ = 2.92 MHz, ηQ = 0.26.
It turns out that the occurrence of carbon in the coordination spheres of silicon and boron depends on the composition of the ceramic rather than on the constitution of a particular single-source precursor. Thus, although the first coordination spheres of silicon and boron in the TSDM polymer still correspond to SiN3C and BN2C, the final ceramic contains silicon and boron exclusively coordinated by nitrogen. However, as soon as the nitrogen content of the polymer decreases, as holds true for the DSDM polymer, carbon is also incorporated into the first coordination spheres of silicon and boron. The main reason behind the preference for the incorporation of nitrogen into the first coordination sphere of the cationic elements rather than carbon can be seen in the higher bonding energies of Si–N and B–N compared to Si–C and B–C. Due to π interactions between nitrogen and silicon, or boron, the energy of the random network is lowered to a larger extent than it would be possible with element–carbon bonds.
2.4 Conclusion
The amorphous silicon boron carbonitrides Si3B3N7C5 and Si3B3N5C7, derived from the new single-source precursors TSDM and DSDM, show a remarkable high-temperature resistance up to 2000 °C, and stability against oxidation in pure oxygen to at least 1300 °C. It has been shown that, due to interconnection of silicon and boron via a methylene bridge, carbon was incorporated into the Si/B/N/C random network at a high degree, leading to notably carbon-rich compositions. The carbon content clearly correlates with the stability of the random networks at high temperatures, which may be ascribed to the fact that carbon bonds to four neighbors instead of three in the case of nitrogen. The first coordination spheres of silicon and boron depend on the composition of the ceramics. While in Si3B3N7C5 silicon and boron are exclusively coordinated by nitrogen (tetrahedrally and trigonal planar, respectively), the carbon-richer compound Si3B3N5C7 exhibits mixed C/N coordination spheres for silicon as well as boron.
By the synthesis of TSDM and DSDM, a new and highly selective synthetic route for the interconnection of silicon and boron via a methylene bridge has been opened. The presented strategy may also be transferred to other cationic elements, opening up a new class of precursor molecules for the preparation of homogeneous multinary ceramics in different systems. The TSDM- and DSDM-derived preceramic polymers are meltable and soluble in organic solvents, thus enabling the preparation of ceramic fibers for CMCs or coatings.
3 Experimental section
3.1 General
All reactions were carried out under inert atmosphere (argon) in rigorously dried reaction apparatus and solvents. Diethyl ether and hexane were distilled from CaH2 (Merck). Commercially available methylamine, trichloroborane (Messer Griesheim), magnesium powder (Riedel–de Haën), triethyl borate (Merck) and chloromethyl trichlorosilane (ABCR) were used without further purification. Chloromethyl dichloro(methyl)silane (kindly provided by the Bayer AG) was distilled under reduced pressure prior to use. IR spectra were recorded between 400 and 4000 cm–1 on a Bruker IFS66 (liquid samples) or on a Bruker IFS 113v (solid samples) FT–IR spectrometer. NMR spectra were recorded using a Bruker Avance DPX-300 SB (liquid and solid samples) and a Bruker Avance DSX-400 WB instrument (solid samples). Mass spectra were measured using a Finnigan TSQ 700 triple stage quadrupole MS. The thermal analyses were carried out in a Netzsch STA 409 (heating rate 10 K min–1) equipped with a Balzers QMS 421 quadrupole MS. The quantitative analysis of nitrogen and oxygen has been carried out simultaneously in a Leco TC-436 hot-gas extraction analyzer, whereas carbon was measured in a Leco C-200 hot-gas extraction analyzer. Silicon and boron were quantified by means of ICP-OES in an Arl 3580 B spectrometer after digestion of the ceramic samples using a mixture of HF/HNO3/HCl.
3.2 Synthesis and spectroscopic data of (C2H5O)2BCl, 1
The synthesis of 1 can be achieved through ligand exchange between triethyl borate and trichloroborane as described in the literature [21,32]. The reaction is carried out by mixing 85.8 g B(OC2H5)3 and 34.6 g BCl3 at –78 °C without any solvent and by stirring the solution at this temperature for 1 h. Distillation of the mixture (51 °C/95 hPa) yielded 112 g of 1.
3.2.1 Spectroscopic data of 1
1H NMR (C6D6): δ = 1.03 (t, 3JHH = 7.03 Hz, O–CH2–CH3), 3.82 (q, 3JHH = 7.03 Hz, O–CH2–CH3). 13C NMR (C6D6): δ = 16.7 (O–CH2–CH3), 62.1 (O–CH2–CH3). 11B NMR (C6D6): δ = 22.9.
3.3 Synthesis and spectroscopic data of Cl3Si–CH2–B(OC2H5), 2 and Cl2(CH3)Si–CH2–B(OC2H5), 3
To a suspension of 7.0 g (288 mmol) magnesium powder in 150 ml dry diethyl ether a few drops of chloromethyl trichlorosilane (chloromethyl dichloro-(methyl)silane) are added. When the Grignard reaction starts (casually gently warming of the reaction flask is necessary) a solution of 36.6 g (199 mmol) chloromethyl trichlorosilane (32.3 g (198 mmol) chloromethyl dichloro(methyl)-silane) in 200 ml diethyl ether is added dropwise to the suspension. The temperature of the mixture has to be kept at 10 °C exactly, and should be controlled very carefully. After adding the silane compound the brown-colored reaction mixture is cooled down to –50 °C and under vigorous stirring 77.8 g (570 mmol) of 1 are added all at once. A few seconds later MgCl2 forms spontaneously, leading to a thick and viscous suspension, which is allowed to reach the room temperature. MgCl2 is filtered off and the filtrate is freed from ether and excess of 1. Distillation of the residue yields 31.3 g (63%) of 2 (b.p. 50 °C/10 hPa) (25.8 g (57%) of 3 (b.p. 62 °C/25 hPa)).
3.3.1 Spectroscopic data of 2
1H NMR (C6D6): δ = 0.87 (s, Si–CH2–B), 1.04 (t, 3JHH = 7.03 Hz, O–CH2–CH3), 3.69 (q, 3JHH = 7.03 Hz, O–CH2–CH3). 13C NMR (C6D6): δ = 12.3 (Si–CH2–B), 17.3 (O–CH2–CH3), 60.2 (O–CH2–CH3). 11B NMR (C6D6): δ = 28.0. 29Si NMR (C6D6): δ = 11.0. MS (EI): m/z (%) 247 (1) [M+–H], 233 (2) [M+–CH3], 219 (2) [M+–C2H5], 213 (3) [M+–Cl], 203 (8) [M+–OC2H5], 175 (25) [203–C2H4], 139 (14) [175–HCl], 101 (100) [B(OC2H5)2+], 73 (60) [B(OC2H5)(OH)+], 63 (6) [SiCl+].
3.3.2 Spectroscopic data of 3
1H NMR (C6D6): δ = 0.64 (s, Si–CH2–B), δ = 0.69 (s, Si–CH3), δ = 1.03 (t, 3JHH = 7.03 Hz, O–CH2–CH3), 3.70 (q, 3JHH = 7.03 Hz, O–CH2–CH3). 13C NMR (C6D6): δ = 7.2 (Si–CH3), δ = 9.3 (Si–CH2–B), 17.4 (O–CH2–CH3), 60.0 (O–CH2–CH3). 11B NMR (C6D6): δ = 28.9. 29Si NMR (C6D6): δ = 31.8. MS (EI): m/z (%) 227 (1) [M+–H], 213 (11) [M+–CH3], 199 (4) [M+–C2H5], 193 (6) [M+–Cl], 185 (26) [213–C2H4], 157 (73) [185–C2H4], 155 (40) [(CH3)Cl2SiCH2B(OH)+], 136 (37) [(CH3)ClSiCHB(OH)2+], 119 (30) [193–O(C2H5)2], 113 (20) [(CH3)C12Si+], 101 (100) [B(OC2H5)2+], 92 (16) [(CH3)ClSiCH2+], 73 (82) [B(OC2H5)(OH)+], 63 (21) [SiCl+].
3.4 Synthesis and spectroscopic data of TSDM, 4 and DSDM, 5
A mixture of 31.3 g (126 mmol) of 2 (25.8 g (113 mmol) of 3) and 42.9 g (366 mmol) trichloroborane is stirred for 30 min at –78 °C. After the solution has reached room temperature, 0.4 g (3 mmol) anhydrous aluminum trichloride is added whereupon the decomposition of the intermediate ethoxy dichloroborane starts slowly and becomes vivid within a few minutes. The evolution of the gaseous by-products can be controlled by occasionally cooling of the reaction mixture. Distillation of the resulting solution yields 24.7 g (85%) of 4 (b.p. 64 °C/48 hPa) (20.6 g (87%) of 5 (b.p. 67 °C/37 hPa)).
3.4.1 Spectroscopic data of 4
1H NMR (C6D6): δ = 1.49 (s). 13C NMR (C6D6): δ = 30.4. 11B NMR (C6D6): δ = 58.6. 29Si NMR (C6D6): δ = 3.1. MS (EI): m/z (%) 228 (2) [M+], 193 (77) [M+–Cl], 157 (43) [193–HCl], 133 (67) [SiCl3+], 121 (12) [157–HCl], 112 (78) [M+–BCl3], 98 (17) [SiCl2+], 81 (24) [BCl2+], 63 (59) [SiCl+], 60 (100) [CH2BCl+]. IR (neat): ν (cm–1) 2957 vw (νas CH2), 2903 w (νs CH2), 1367 s (γ CH2), 1121 vs (νas B–C), 887 vs (νas BCl2), 748 vs (νas Si–C), 621 vs (νas SiCl3).
3.4.2 Spectroscopic data of 5
1H NMR (C6D6): δ = 0.47 (s, Si–CH3), δ = 1.47 (s, Si–CH2–B). 13C NMR (C6D6): δ = 6.5 (Si–CH3), δ = 29.3 (Si–CH2–B). 11B NMR (C6D6): δ = 59.2. 29Si NMR (C6D6): δ = 23.9. MS (EI): m/z (%) 208 (1) [M+], 193 (32) [M+–CH3], 173 (20) [M+–Cl], 157 (33) [193–HCl], 137 (11) [173–HCl], 121 (2) [157–HCl], 113 (100) [(CH3)Cl2Si+], 98 (3) [SiCl2+], 92 (4) [M+–BCl3], 81 (11) [BCl2+], 63 (32) [SiCl+], 60 (34) [CH2BCl+]. IR (neat): ν (cm–1) 2974 w (νas CH2/CH3), 2902 w (νs CH2/CH3), 1366 s (γ CH2), 1117 vs (ν B–C), 885 vs (νas BCl2), 792 s (ν Si–C), 759 s (ν Si–C), 595 s (νas SiCl2).
3.5 Polycondensation of 4 and 5 with methylamine
A solution of 13.9 g (61 mmol) 4 or 11.7 g (56 mmol) 5 in 150 ml hexane is added dropwise to a solution of 100 ml methylamine in 150 ml hexane at –78 °C. The stirred mixture is allowed to reach room temperature overnight where upon excessive methylamine evaporates. After filtration of the accumulated methylammonium hydrochloride the residue is washed two times with 50 ml hexane. Hexane is removed from the filtrate whereupon the polymer remains as a viscous colorless liquid, which is annealed at 80 °C for 24 h.
3.6 Pyrolysis of the polyborocarbosilazanes
The polymers derived from 4 and 5 are pyrolyzed in a first step up to 900 °C under flowing argon in a boron nitride crucible. The heating starts at room temperature at a rate of 100 K h–1 up to 300 °C, followed by a dwell of 3 h to complete cross linking of the polymer. After that, the temperature is raised to 900 °C at 100 K h–1 and maintained for another 3 h. Finally, the pyrolyzed samples are heated up to 1500 °C at 300 K h–1 in an alumina crucible under argon and are calcinated at this temperature for 3 h.