

1 Introduction
The activation of single or multiple C–N bonds plays a relevant role to understand the mechanism involved in the hydrodenitrogenation of fossil fuels to reduce pollution [1]. Furthermore, the catalytic hydrogenation of organic substrates containing a multiple C≡N, or C=N bond is an important process to produce a variety of amines such as benzylamine, 3-alkoxypropylenamines, hexamethylendiamine, etc. [2]. Amines or their derivatives have a great range of applications such as solvents, agrochemicals [3], pharmaceuticals [4] and others [5].
The catalytic hydrogenation of nitrile and azo-compounds under a high pressure of hydrogen was realized using supported catalysts [6,7] or in the presence of transition metal complexes in homogeneous phase [8,9,10]. Many examples of heterogeneous hydrogenations are known, and particular attention was given to the use of Pd or other transition metals supported on carbon, alumina or silica, as catalyst [11]. Rare are the reports of the process carried out in homogeneous phase, sometimes dedicated to imine hydrogenation [12], as intermediates of nitrile hydrogenation. The products are the same, independently of the heterogeneous or homogeneous process: a mixture of primary, secondary, and sometimes tertiary amines were obtained in the hydrogenation of nitriles [6,7]. Winans and Adkins [13] reported a mechanism to explain the products formed when nitriles where reduced: a first hydrogenation of C≡N gives the corresponding imine that, in a subsequent step, is hydrogenated to the primary amine. This reduction is followed by a coupling reaction between the intermediate imine and the amine to give, in the presence of hydrogen, the secondary amine and ammonia. The secondary amine through an analogous reaction path may give the tertiary amine and ammonia in a further step.
In this paper, we report the catalytic activity of three Ru(II) dihydride complexes in the homogeneous hydrogenation of R–C≡N and R–CH=N–R compounds. The RuH2(CO)2(PnBu3)2 (1), RuH2(CO)2(PPh3)2 (2), RuH2(PPh3)4 (3) complexes were chosen in consideration of their well-known catalytic activity in the hydrogenation of unsaturated substrates such as alkenes and carbonylic compounds [14]. The influence of ancillary ligand coordinated to ruthenium (II) complexes, having different electronic or steric effect, were evaluated.
The different reaction parameters such as reaction time, temperature, hydrogen pressure and catalyst/substrate ratio were tested. The activation parameters and the kinetic constants for these reactions were also calculated to collect information about the mechanism involved.
2 Experimentals
2.1 Instruments and materials
A Shimadzu GC14 chromatograph equipped with two FID detectors, using 2-m packed columns filled with a FFAP (free fatty acid phase) or CW 20 M + KOH as stationary phase was used for quantitative analyses. p-Xylene was employed as internal standard. The response factors of reagents and products versus p-xylene were evaluated. The identity of the products was confirmed by GC–MS using a Shimadzu apparatus (GCMS-QP5050A) equipped with a capillary column SPtm-1 (length 30 m, diameter 0.25 mm, film thickness 0.1 μm).
Elemental analyses were performed with a Perkin-Elmer Analyser model 2400 Series II CHNS/O.
IR spectra were recorded with a Perkin-Elmer mod. 1760 FTIR spectrometer.
1H, 13C and 31P NMR spectra were recorded using a Varian VXR300 spectrometer operating at 299.987 MHz for 1H, at 75.429 MHz for 13C and at 121.421 MHz for 31P NMR, using solutions in appropriate solvents. SiMe4 was used as external standard for 1H and 13C NMR, H3PO4 (85%) for 31P NMR (signals reported as positive downfield to the standard). 13C and 31P NMR spectra were acquired using a broadband decoupler.
All manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques.
Hydrogenations were performed in a Parr high-pressure autoclave (150 ml) or in a glass vial inserted in a homemade high-pressure vessel (150 ml).
Tetrahydrofuran (THF) was dried and deoxygenated by refluxing and distilling over sodium/potassium amalgam under nitrogen atmosphere.
The following catalysts were prepared according to the literature and their spectroscopic characteristics were in agreement with the data reported: Ru(CO)2(CH3COO)2(PnBu3)2 [15], Ru(CO)2(CH3COO)2(PPh3)2 [16], RuH2(CO)2(PnBu3)2 (1) [17], RuH2(CO)2(PPh3)2 (2) [18], RuH2(PPh3)4 (3) [19].
Benzonitrile: the commercial product was distilled under nitrogen at reduced pressure before use (b.p.: 80 °C/1 mmHg).
N-Benzylidenaniline: the commercial product was employed as received.
2.2 Hydrogenation experiments
A typical hydrogenation experiment was reported for each ruthenium catalyst: the same procedure was employed for all substrates and the data collected on the influence of temperature, hydrogen pressure, catalyst concentration and reaction time were reported in Tables 1–4.
Reduction of benzonitrile in the presence of RuH2(CO)2(PnBu3)2 (1), RuH2(CO)2(PPh3)2 (2), and RuH2(PPh3)4 (3)
| Entry | Cat. code | T (K) | H2 (atm) | time (h) | [Cat] (mM) | Conv. (%) | Reaction mixture composition (%) | ||
| (I) | (II) | (III) | |||||||
| 1 | 1 | 373 | 50 | 3 | 0.66 | 8.7 | 94.3 | 2.4 | 3.3 |
| 2 | 1 | 383 | 50 | 3 | 0.66 | 18.4 | 88.4 | 3.3 | 8.3 |
| 3 | 1 | 393 | 50 | 3 | 0.66 | 32.6 | 79.3 | 3.0 | 17.7 |
| 4 | 1 | 393 | 50 | 6 | 0.66 | 36.6 | 75.9 | 4.3 | 19.8 |
| 5 | 1 | 393 | 50 | 9 | 0.66 | 43.3 | 69.3 | 8.4 | 22.3 |
| 6 | 1 | 393 | 50 | 24 | 0.66 | 55.8 | 57.6 | 12.1 | 30.3 |
| 7 | 1 | 393 | 25 | 3 | 0.66 | 4.1 | 97.1 | 1.6 | 1.3 |
| 8 | 1 | 393 | 75 | 3 | 0.66 | 52.9 | 59.1 | 15.5 | 25.4 |
| 9 | 1 | 393 | 50 | 3 | 0.23 | 17.5 | 89.7 | 1.5 | 8.8 |
| 10 | 1 | 393 | 50 | 3 | 1.42 | 48.5 | 62.3 | 16.8 | 20.9 |
| 11 | 2 | 363 | 50 | 3 | 0.66 | 15.1 | 91.7 | 0.3 | 8.0 |
| 12 | 2 | 383 | 50 | 3 | 0.66 | 26.4 | 84.6 | 0.5 | 14.9 |
| 13 | 2 | 393 | 50 | 3 | 0.66 | 49.7 | 66.6 | 1.0 | 32.4 |
| 14 | 2 | 393 | 50 | 6 | 0.66 | 57.6 | 57.2 | 7.9 | 34.9 |
| 15 | 2 | 393 | 50 | 9 | 0.66 | 66.9 | 45.4 | 17.5 | 37.1 |
| 16 | 2 | 393 | 50 | 24 | 0.66 | 76.5 | 31.9 | 32.6 | 35.5 |
| 17 | 2 | 393 | 25 | 3 | 0.66 | 19.7 | 87.9 | 2.6 | 9.5 |
| 18 | 2 | 393 | 75 | 3 | 0.66 | 57.0 | 59.0 | 3.8 | 37.2 |
| 19 | 2 | 393 | 50 | 3 | 0.23 | 42.5 | 72.7 | 0.8 | 26.5 |
| 20 | 2 | 393 | 50 | 3 | 1.42 | 51.2 | 64.6 | 3.0 | 32.4 |
| 21 | 3 | 363 | 50 | 3 | 0.66 | 19.2 | 89.3 | 0.2 | 10.5 |
| 22 | 3 | 383 | 50 | 3 | 0.66 | 38.5 | 76.0 | 0.5 | 23.5 |
| 23 | 3 | 393 | 50 | 3 | 0.66 | 50.3 | 66.1 | 0.9 | 33.0 |
| 24 | 3 | 393 | 50 | 6 | 0.66 | 55.2 | 61.8 | 0.1 | 38.1 |
| 25 | 3 | 393 | 50 | 9 | 0.66 | 66.5 | 46.8 | 13.3 | 39.9 |
| 26 | 3 | 393 | 50 | 24 | 0.66 | 72.4 | 39.2 | 18.7 | 42.1 |
| 27 | 3 | 393 | 10 | 3 | 0.66 | 15.8 | 91.4 | 0 | 8.6 |
| 28 | 3 | 393 | 25 | 3 | 0.66 | 31.2 | 81.5 | 0 | 18.5 |
| 29 | 3 | 393 | 75 | 3 | 0.66 | 68.4 | 47.7 | 1.5 | 50.8 |
| 30 | 3 | 393 | 50 | 3 | 0.23 | 48.7 | 67.8 | 0 | 32.2 |
| 31 | 3 | 393 | 50 | 3 | 1.42 | 51.2 | 63.4 | 6.8 | 29.8 |
Kinetic and thermodynamic data for the reduction of benzonitrile in the presence of RuH2(CO)2(PnBu3)2 (1), RuH2(CO)2(PPh3)2 (2), and RuH2(PPh3)4 (3)
| Ru(II) complex | code | Kc × 106 | R2 | Kp × 106 | R2 | Kcat × 106 | R2 | ΔH** | R2 | ΔS** | R2 |
| (s–1) | (s–1 atm–1) | (s–1 mmol–1 l) | (kJ mol–1) | (J mol–1 K–1) | |||||||
| RuH2(CO)2(PnBu3)2 | (1) | 5.5 | 0.98 | 1.3 | 1.00 | 36.1 | 0.99 | 86.1 | 0.99 | –113 | 0.99 |
| RuH2(CO)2(PPh3)2 | (2) | 9.4 | 0.92 | 1.2 | 0.92 | 11.7 | 0.76 | 51.0 | 0.96 | –199 | 0.96 |
| RuH2(PPh3)4 | (3) | 7.3 | 0.83 | 1.4 | 0.99 | 3.7 | 0.83 | 44.1 | 1.00 | –216 | 1.00 |
Hydrogenation of N-benzylidenaniline in the presence of RuH2(CO)2(PnBu3)2 (1), RuH2(CO)2(PPh3)2 (2), and RuH2(PPh3)4 (3)
| Entry | Cat | T | H2 | r.t. | [Cat] | Conv. | Reaction mixture composition (%) | |||
| N. | code | (K) | (atm) | (h) | (mM) | (%) | (V) | (VI) | (VII) | (VIII) |
| 32 | 1 | 363 | 50 | 3 | 0.66 | 3.0 | 96.0 | 2.0 | 1.0 | 1.0 |
| 33 | 1 | 383 | 50 | 3 | 0.66 | 3.7 | 95.1 | 2.5 | 1.2 | 1.2 |
| 34 | 1 | 393 | 50 | 3 | 0.66 | 5.8 | 92.0 | 3.4 | 2.3 | 2.3 |
| 35 | 1 | 393 | 50 | 6 | 0.66 | 16.1 | 77.0 | 6.6 | 8.2 | 8.2 |
| 36 | 1 | 393 | 50 | 9 | 0.66 | 27.7 | 59.7 | 5.5 | 17.4 | 17.4 |
| 37 | 1 | 393 | 50 | 24 | 0.66 | 50.1 | 34.6 | 4.2 | 30.6 | 30.6 |
| 38 | 1 | 393 | 25 | 3 | 0.66 | 1.6 | 97.8 | 1.0 | 0.6 | 0.6 |
| 39 | 1 | 393 | 75 | 3 | 0.66 | 14.9 | 79.7 | 7.5 | 6.4 | 6.4 |
| 40 | 1 | 393 | 50 | 3 | 0.23 | 3.8 | 94.5 | 1.9 | 1.8 | 1.8 |
| 41 | 1 | 393 | 50 | 3 | 1.42 | 13.1 | 79.9 | 3.9 | 8.1 | 8.1 |
| 42 | 2 | 363 | 50 | 3 | 0.66 | 4.4 | 93.9 | 2.5 | 1.8 | 1.8 |
| 43 | 2 | 383 | 50 | 3 | 0.66 | 5.5 | 90.5 | 1.1 | 4.2 | 4.2 |
| 44 | 2 | 393 | 50 | 3 | 0.66 | 7.6 | 86.2 | 0.4 | 6.7 | 6.7 |
| 45 | 2 | 393 | 50 | 6 | 0.66 | 16.3 | 73.0 | 1.4 | 12.8 | 12.8 |
| 46 | 2 | 393 | 50 | 9 | 0.66 | 34.2 | 52.6 | 7.2 | 20.1 | 20.1 |
| 47 | 2 | 393 | 50 | 24 | 0.66 | 82.6 | 10.4 | 9.4 | 40.1 | 40.1 |
| 48 | 2 | 393 | 25 | 3 | 0.66 | 5.9 | 91.0 | 2.4 | 3.3 | 3.3 |
| 49 | 2 | 393 | 75 | 3 | 0.66 | 15.8 | 73.2 | 0.6 | 13.1 | 13.1 |
| 50 | 2 | 393 | 50 | 3 | 0.23 | 5.1 | 92.0 | 1.8 | 3.1 | 3.1 |
| 51 | 2 | 393 | 50 | 3 | 1.42 | 19.3 | 68.6 | 1.4 | 15.0 | 15.0 |
| 52 | 3 | 373 | 50 | 3 | 0.66 | 14.5 | 82.3 | 10.3 | 3.7 | 3.7 |
| 53 | 3 | 383 | 50 | 3 | 0.66 | 25.7 | 69.2 | 17.0 | 6.9 | 6.9 |
| 54 | 3 | 393 | 50 | 3 | 0.66 | 42.5 | 51.3 | 27.1 | 10.8 | 10.8 |
| 55 | 3 | 393 | 50 | 6 | 0.66 | 53.6 | 39.3 | 30.1 | 15.3 | 15.3 |
| 56 | 3 | 393 | 50 | 9 | 0.66 | 64.3 | 29.0 | 33.4 | 18.8 | 18.8 |
| 57 | 3 | 393 | 50 | 24 | 0.66 | 92.1 | 5.8 | 40.4 | 26.9 | 26.9 |
| 58 | 3 | 393 | 25 | 3 | 0.66 | 42.0 | 52.8 | 29.4 | 8.9 | 8.9 |
| 59 | 3 | 393 | 75 | 3 | 0.66 | 43.4 | 49.5 | 25.5 | 12.5 | 12.5 |
| 60 | 3 | 393 | 50 | 3 | 0.23 | 40.4 | 58.6 | 38.2 | 1.6 | 1.6 |
| 61 | 3 | 393 | 50 | 3 | 1.42 | 42.9 | 44.5 | 11.3 | 22.1 | 22.1 |
Kinetic and thermodynamic data for the reduction of N-benzylidenaniline in the presence of RuH2(CO)2(PnBu3)2 (1), RuH2(CO)2(PPh3)2 (2), and RuH2(PPh3)4 (3)
| Ru(II) complex | code | Kc × 106 | R2 | Kp × 108 | R2 | Kcat × 106 | R2 | ΔH** | R2 | ΔS** | R2 |
| (s–1) | (s–1 atm–1) | (s–1 mmol–1 l) | (kJ mol–1) | (J mol–1 K–1) | |||||||
| RuH2(CO)2(PnBu3)2 | (1) | 8.1 | 0.98 | 26.8 | 0.95 | 8.1 | 0.98 | 21.5 | 0.99 | –293 | 0.99 |
| RuH2(CO)2(PPh3)2 | (2) | 23.0 | 0.99 | 21.5 | 0.90 | 13.1 | 0.96 | 18.0 | 0.99 | –300 | 0.99 |
| RuH2(PPh3)4 | (3) | 26.7 | 1.00 | 4.5 | 0.97 | 3.0 | 0.74 | 73.8 | 1.00 | –142 | 1.00 |
2.2.1 Hydrogenation in the presence of RuH2(CO)2(PnBu3)2 (1) or RuH2(CO)2(PPh3)2 (2)
The catalyst was prepared immediately before its use from the corresponding acetato complex, following the procedure reported by Salvini [17] for (1) or Frediani [18] for (2).
Ru(CO)2(CH3COO)2(PR3)2 (1.32 × 10–5 mol), 4 ml of anhydrous THF, and Na2CO3 (0.5 g) were introduced in a glass vial inserted in an autoclave, then hydrogen up to 100 bar was added. The vessel was heated at 100 °C for 14 h in a thermostated oil bath. The reactor was cooled, the gas vented out and the yellow solution filtered and transferred in a Schlenk tube containing the substrate (1.32 × 10–3 mol) and p-xylene (160 μl) as internal standard. A further amount of THF was added up to a total volume of 20 ml. The solution was introduced in a high-pressure vessel under nitrogen atmosphere, then hydrogen up to the pressure required was added. The vessel was rocked in an oil bath, heated at the prefixed temperature, for the time required.
At the end of the reaction the vessel was cooled down, the gas vented out and the solution analysed by GC and GC–MS techniques.
2.2.2 Hydrogenation in the presence of RuH2(PPh3)4 (3)
In a Schlenk tube RuH2(PPh3)4 (1.32 × 10–5 mol), substrate (1.32 × 10–3 mol) and p-xylene (160 μl) as internal standard were dissolved in THF (20 ml).
The solution was transferred by suction in a Parr autoclave and after addition of hydrogen up to the pressure required, the reactor was stirred at the established temperature.
At the end of the reaction, the vessel was cooled down, the gas vented out and the solution analysed by GC and GC–MS techniques.
3 Results and discussion
3.1 Hydrogenation of benzonitrile
In the hydrogenation of benzonitrile (I), two different products were obtained: benzylamine (II) and N,N-dibenzylamine (III) (Scheme 1). Taking into account the mechanism reported by Winans and Adkins [10], the reduction of benzonitrile takes place with formation of benzylimine (IV) that was subsequently hydrogenated to benzylamine (II). The secondary amine is formed by a coupling reaction between (II) and (IV) to give, in the presence of hydrogen, dibenzylamine (III) and NH3. The reaction of (III) with (IV) to give the tertiary amine does not take place. The results of the hydrogenation of (I) are reported in Table 1.
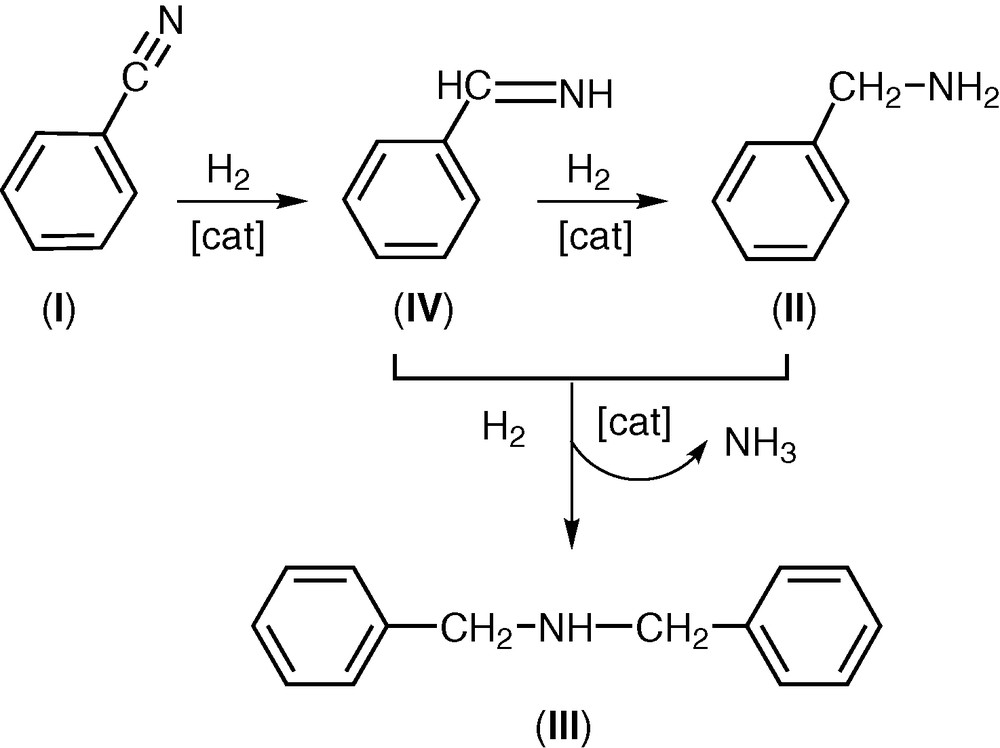
Hydrogenation of benzonitrile.
The catalyst (1) is catalytically active at 363 K, but the conversion is only 8.7% after 3 h (entry 1, Table 1). Rising the temperature from 363 to 393 K, the conversion increases up to 32.6% (entries 1–3). Furthermore, an increase of the reaction time up to 24 h at 393 K increases the conversion of (I) up to 55.8% (entries 3–6). An analogous beneficial effect is shown by an increment of hydrogen pressure: increasing the pressure from 25 to 75 atm, the conversion rises from 4.1% to 52.9% (entries 7, 3 and 8).
Furthermore, the conversion rises from 17.5 to 48.5% when the concentration of (1) increases from 0.23 to 1.42 mM (entries 9, 3 and 10).
An analysis of the reaction products obtained in the hydrogenation of (I) using the catalyst (1) shows that the main product is always N,N-dibenzylamine (III), while an appreciable amount of benzylamine (II) (concentration 3.0%) is present after 3 h (entry 3) and its concentration rises up to 12.1% after 24 h (entry 6).
Catalyst (2) shows a conversion of 15.1% working at 363 K after 3 h under a hydrogen pressure of 50 atm (entry 11), but increasing the temperature from 363 to 393 K the conversion rise up to 49.7% (entry 13). When the reaction time is prolonged from 3 up to 24 h, the conversion of (I) increases up to 76.5% (entries 13–16).
A beneficial effect of the hydrogen pressure is shown in the hydrogenation of (I), since the conversion increases from 19.7 to 57.0% when the hydrogen pressure increases from 25 up to 75 atm (entries 17, 13, and 18).
Also an increment of the catalyst concentration from 0.23 to 1.42 mM rises the conversion from 42.5 to 51.2% (entries 19, 13, and 20).
The main product is always N,N-dibenzylamine (III); however, N-benzylamine (II) is present in all tests.
The catalyst (3) hydrogenates benzonitrile (I) to give the N,N-dibenzylamine (III) as the main product, with a conversion of 19.2% at 363 K (entry 21). Increasing the temperature from 363 to 393 K, the conversion of (I) rises from 19.2% up to 50.3% and the main product still remains the N,N-dibenzylamine (III) (entries 21–23, Table 1).
Also an increase of the reaction time from 3 to 24 h at 393 K rises the conversion up to 72.4% (entries 23–26). In these tests, (III) is always the main product, but an appreciable concentration of N-benzylamine (II) is present after 9 h (13.3%, entry 25), and rises up to 18.7% after 24 h.
Also an increase of the hydrogen pressure from 10 up to 75 atm increases the conversion from 15.8 up to 68.4% (entries 27, 28, 23 and 29) after a reaction time of 3 h at 393 K.
The concentration of (3) shows a slight beneficial effect, in fact, an increase of catalyst concentration from 0.23 up to 1.42 mM raises the conversion from 48.7 to 51.2% (entries 30, 23, and 31).
Kinetic analyses of the data reported in Table 1, following the procedure reported by Salvini et al. [14], shows a first partial order with respect to reaction time, hydrogen pressure and catalyst concentration with all the catalysts tested.
The kinetic rates, with respect to reaction time are not very different with values of 5.5 × 10–6 s–1 for (1), 9.4 × 10–6 s–1 for (2) and 7.3 × 10–6 s–1 for (3) (Table 2). The influence of hydrogen pressure on the initial rate (Kp) is almost the same, with values in the range 1.2–1.4 × 10–6 s–1 atm–1, while the changes of the catalyst concentration affect the initial rate (Kcat) in the following order: (1) > (2) > (3).
The activation parameters were also evaluated [20] and the values of the activation entropy, with all catalysts, are negative, indicating an associative process in the rate-determining step of the reaction (Table 2).
3.2 Hydrogenation of N-benzylidenaniline
In the hydrogenation of N-benzilidenaniline (V), three different products were obtained, that is N-benzylaniline (VI), due to the hydrogenation of the C=N group, aniline (VII) and toluene (VIII): they are the products of the hydrogenolisis of the CH2–NHPh bond of N-benzylaniline (Scheme 2).

Reduction of N-benzylidenaniline.
The results of the reduction of this substrate with the three different Ru(II) complexes (1)–(3) are reported in Table 3.
As we can see, (V) is not able to condensate with a molecule of (VI), in the reaction conditions tested. This behaviour is different from that shown in the reduction of (I) where, as reported above, the reaction of (IV) with (II) gives (III) as the main product. The absence of this reaction may be ascribed to the higher rigidity of (V) with respect to (IV) or to the high steric hindrance of the phenyl group linked to the nitrogen atom. Furthermore, (III) is not hydrogenolysed to (II) and (VIII), while in the reduction of (V) the formation of (VII) and (VIII) were shown. The different behaviour between (I) and (V) in the catalytic hydrogenation may be ascribed to the different steric and electronic properties of (III) and (VI). The nitrogen atom in (VI) is directly linked to an aromatic carbon atom, while in (III) it is linked to an aliphatic carbon atom. This hypothesis is confirmed by the products detected in the hydrogenolysis of (VI). In fact, (VII) and (VIII) were present as reaction products, while benzene and benzylamine was never identified in this reaction mixture.
The catalyst (1) displays a low activity in the hydrogenation of (V) and only a conversion of 3.0% was obtained after 3 h at 363 K under 50 atm of hydrogen pressure (entry 32). A moderate increment of conversion up to 5.8% is shown when the temperature rises up to 393 K (entries 32–34). Also an increase of the conversion from 1.6 up to 14.9% was obtained when the hydrogen pressure rises from 25 up to 75 atm (entries 38, 34 and 39). However working for 24 h at 393 K under 50 atm of hydrogen, a 50.1% conversion has been obtained (entry 37).
Furthermore, the conversion increases from 3.8% to 13.1% when the concentration of the catalyst increases from 0.23 to 1.42 mM (entries 40, 34 and 41).
A slightly higher activity is shown by catalyst (2) with a conversion of 4.4%, working with 50 atm of hydrogen at 363 K for 3 h, which increases up to 7.6% when the temperature rises up to 393 K (entries 42–44). In the same conditions, an 82.6% conversion is obtained if the reaction is prolonged to 24 h (entry 47).
A higher influence of the hydrogen pressure is shown with respect to that reported when (1) is employed, the conversion increases from 5.9 to 15.8% when the hydrogen increases from 25 to 75 atm (entries 48, 44 and 49). Furthermore an increment of the catalyst concentration from 0.23 to 1.42 mM causes an increase in the conversion from 5.1% up to 19.3% (entries 50, 44 and 51).
The higher catalytic activity is shown by (3) with a conversion of 14.5% at 373 K, which increases up to 42.5% (entries 52–54) when the temperature rises to 393 K. The rise in the hydrogen pressure from 25 to 75 atm shows a very low increase in the conversion from 42.0 to 43.4% (entries 58, 54 and 59), while working for 24 h at 393 K, in the presence of 50 atm of hydrogen (entry 57), a 92.1% conversion is obtained.
Employing the data reported in Table 3, we have evaluated that the conversion of N-benzylidenaniline shows a first partial order rate with respect to reaction time, hydrogen pressure and catalyst concentration with all the catalysts tested. The catalyst (2) and (3) show a similar Kc (respectively 2.30 × 10–5 s–1 and 2.67 × 10–5 s–1), while (1) is less affected by substrate concentration (Kc 8.1 × 10–6) (Table 4). On the contrary, the specific rates with respect to the hydrogen pressure (Kp) follow the order (1) > (2) > (3), while those related to the catalyst concentration (Kcat) are in the order (2) > (1) > (3).
The thermodynamic values of the activation parameters were also calculated (Table 4) for these catalysts. The activation entropies are always negative, in agreement with an associative process involved in the rate-determining step.
3.3 On the mechanism of benzonitrile hydrogenation
The thermodynamic data collected (Table 2) suggest that the same rate-determining step is involved using catalyst (1)–(3). In fact, the negative value of ΔS** suggests an approach to the transition state involving an increase of the steric hindrance. Taking into account that (3) shows the higher catalytic activity and it easily dissociates a phosphine ligand, we propose a first step where a phosphine is displaced by a benzonitrile, forming the complex (A) (Scheme 3).
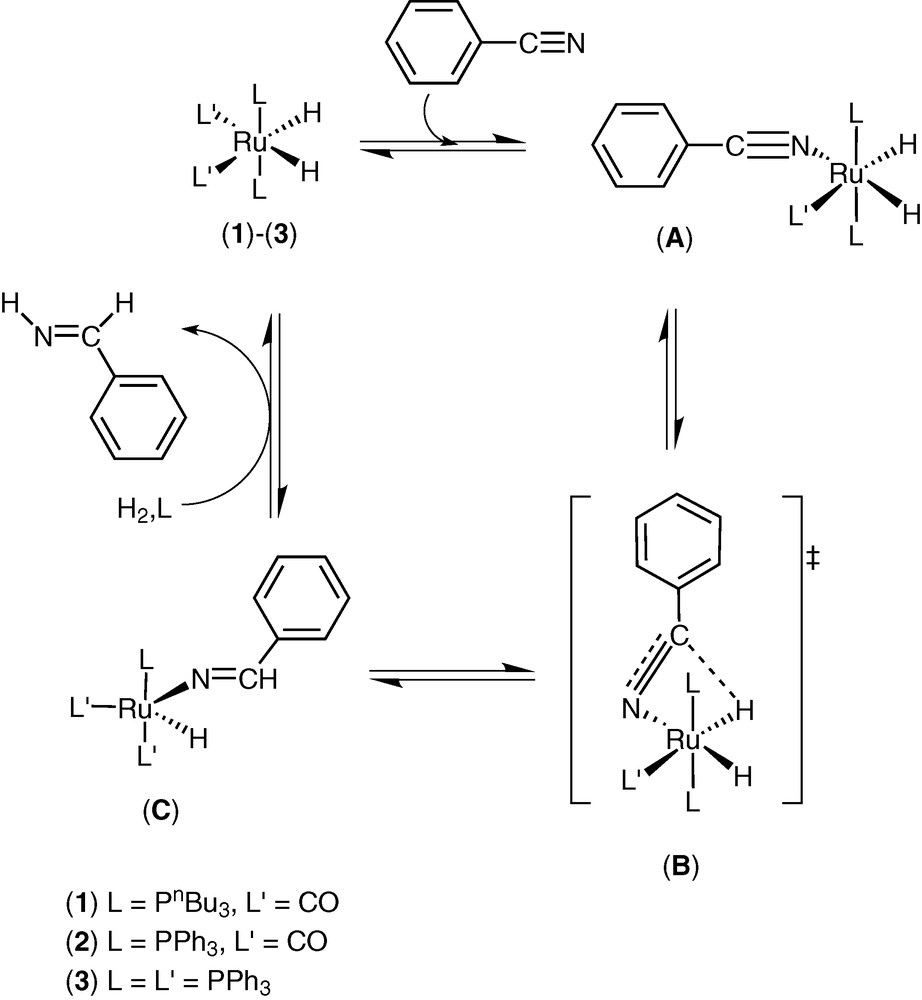
The following step is the insertion of C≡N bond into a Ru–H through a four-centre intermediate (B) forming an imino Ru complex (C). The hydrogenolysis of this complex gives the imine (IV) and restores the catalyst (3). We assume that an analogous process is involved in the reduction of (IV) to give the benzylamine (II), that is a dihydroimino complex is formed, followed by the insertion of a C=N bond into a Ru–H, obtaining an aminoruthenium complex. The benzylamine (II) is formed through hydrogenolysis of the aminoruthenium complex and the catalyst (3) is restored.
This hypothesis on the mechanism of benzonitrile hydrogenation is supported by the preliminary data on the reactivity of the complex (3) with benzonitrile (I). A solution of (3) in hexadeuterobenzene reacts with (I) at 25 °C, giving a new complex (A) (L, L′ = PPh3) characterized by 1H, 31P NMR and IR spectroscopies (Fig. 1). In the hydride region of 1H NMR (300 MHz) there is a spin system ABX2Y with signals at δ = –13.79 (tdd, 1H, HA, JHAPA = 28.44 Hz, JHAPB = 13.77 Hz, JHAHB = 6.80 Hz), –8.57 ppm (dtd, HB, JHBPB = 77.17 Hz, JHBPA = 30.85 Hz, JHBHA = 6.80 Hz); the 31P–{1H} NMR (121.4 MHz) shows signals at δ = 48.1 (t, 1P, PB, JPBPA = 16.51 Hz), 61.2 (d, 2P, PA, JPAPB = 16.51 Hz); IR νCN 2200 cm–1. An analogous 1H NMR spin system was reported for RuH2(N2)(PPh3)3 [21].
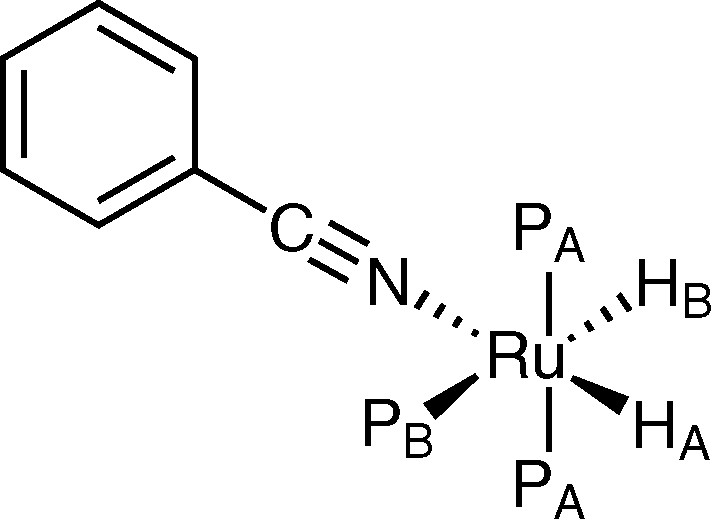
Hypothesised structure of complex (A).
Heating the solution at 80 °C for 24 h, two new complexes were formed (C) and (C′) (L, L′ = PPh3) in a 11/1 ratio, characterized by 1H, and 31P NMR spectroscopy (Fig. 2). Complex (C) shows a spin system A2BX with signals in the hydride region of 1H NMR (300 MHz) at δ = –18.97 ppm (dt, 1H, JHPA = 25.51 Hz, JHPB = 24.06 Hz); a further signal was present at δ = 7.88 (br, 1H, CH=N) ppm, attributed to CH of the imine group; the 31P NMR shows signals at δ = 48.1 (d, 2P, JPAPB = 27.56 Hz), 68.6 (t, 1P, JPAPB = 27.56 Hz). The IR spectrum shows a νC=N at 1726 cm–1. An alternative square pyramidal structure with the hydride in the apical position may be hypothesised for (C), taking into account the very high field of the hydride resonance, as reported for some transition metal complexes [22] and suggested by one of the referees.
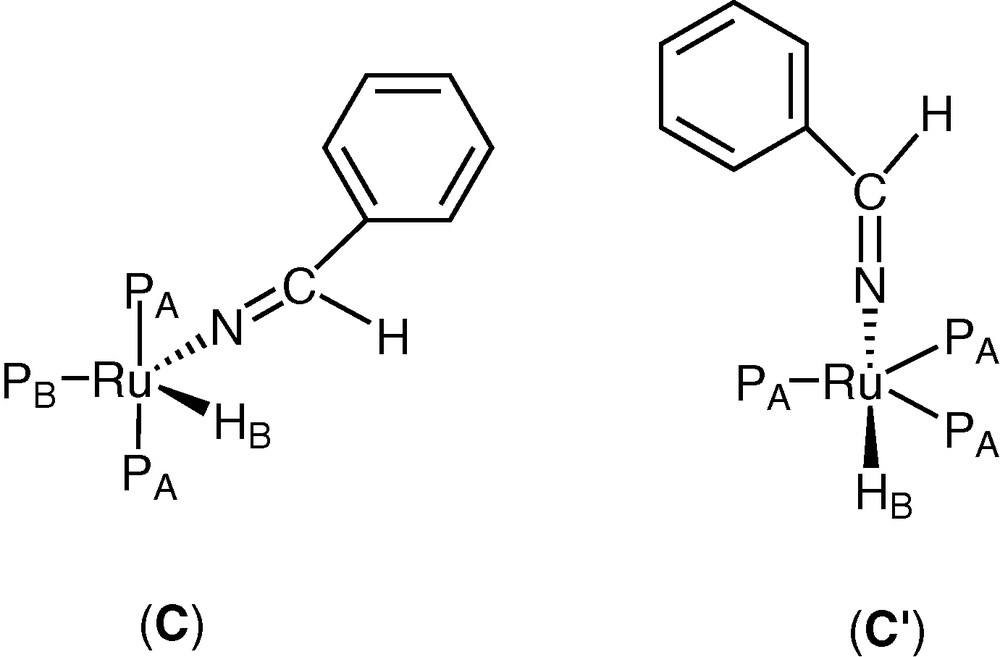
Hypothesised structures of complexes (C) and (C′).
An A3X spin system was present for complex (C′) in the hydride region of 1H NMR at δ = –17.72 ppm (q, 1H, JHP = 25.91 Hz), while the resonance of the CH of the imine group was not evidenced, due to the low concentration of this complex; the 31P NMR shows a singlet at δ = 35.8.
Analogous reaction paths may be hypothesised for the hydrogenation of benzylidenaniline (V) to benzylaniline (VI) and its hydrogenolysis to aniline (VII) and toluene (VIII).
4 Conclusion
In the hydrogenation of nitriles, the complex having the greater activity is RuH2(CO)2(PPh3)2 (2), with a conversion up to 76.5% after 24 h at 393 K. Two different products are formed, benzylamine (II) (from the reduction of benzonitrile (I)) and N,N-dibenzylamine (III) (through the condensation of benzylamine (II) and benzylimmine (IV)).
An appreciable amount of benzylamine is already present after reaction times of 6 and 9 h (entries 14–15 or 24–25) and the increase in benzylamine concentration may be attributed to the negative influence of free NH3 present in solution and formed by the reaction between (IV) and (II). It is known that free ammonia inhibits the condensation, according to the mechanism reported by Winans et al. [10].
These catalysts are also active in the hydrogenation of a C=N double bond and in the hydrogenolisis of the C–N bond. The conversion of N-benzylidenaniline (V) is 92.1% after 24 h at 393 K in the presence of (3). The hydrogenation of the C=N double bond forms in a first step the corresponding N-benzylaniline (VI). When an appropriate amount of N-benzylaniline is present in the reaction medium, aniline and toluene are the main products, as shown after a reaction time of 24 h (entries 57, 47, 37).
The data collected show that (1) the catalyst containing the more basic phosphine is less active, in agreement with a dissociation of the phosphine in the first step of the catalytic cycle. However, the substitution of two carbonyl groups in (2) with two triphenylphosphine ligands gives (3) a catalyst having almost the same activity.
The thermodynamic data collected are in agreement with an associative mechanism in the rate-determining step of this reaction, independently of the catalysts tested. The ability of these catalysts in the hydrogenolysis of a C–N single bond opens the possibility of using them in the hydrodenitrogenation of fossil fuels or of other nitrogen-containing compounds.
Acknowledgements
The authors thank the University of Florence, and the ‘Ministero della Industria, Università e Ricerca’ (MIUR), the ‘Programmi di Ricerca Scientifica di Notevole Interesse Nazionale, Cofinanziamento MIUR 2003-04’, for financial support. We also thank Brunella Innocenti and Maurizio Passaponti, Department of Organic Chemistry, University of Florence, for elemental analyses.