

1 Introduction
Transition-metal salts or complexes catalyse the electrosynthesis of dialkyl carbonate from alcohols and carbon monoxide [1] including Palladium salts or complexes [2,3] (Eq. (1)). This anodic process proceeds in methanol or ethanol under mild conditions i.e., at room temperature and with CO at atmospheric pressure, leading to dimethyl carbonate (DMC) [2] and diethyl carbonate (DEC) respectively [3].
| (1) |
This process is an alternative to procedures involving chemical oxidants such as oxygen [4–6], which generally require more drastic conditions (high temperatures and high CO pressures) or electrolytic carbonylation in the gas phase [7,8]. The mechanism of alcohols carbonylation involving stoichiometric amounts of PdII complexes has been reported. It proceeds via the formation of intermediate alkoxycarbonyl complexes XPdII(COOR)L2 (X = Cl, OAc; L = PPh3, dppe; R = Me, Et) obtained from PdII complexes under high CO pressures and high temperatures. In the presence of a base (trialkylamine) and alcohol, they evolve to dialkyl carbonates and Pd0 complexes [9–12]. The process is not catalytic in the absence of any oxidants.
We report here a mechanism for the electrosynthesis of diethyl carbonate from ethanol and CO catalysed
| (2) |
The electrosynthesis was more efficient in the presence of catalytic amounts of PdBr2 [2,3]. However, the reduction of PdBr2 observed by cyclic voltammetry resulted in poor resolved voltammograms due to a lack of stabilization of the electrogenerated Pd0 that deposited at the electrode. Consequently the mechanism has been investigated with PdBr2(PPh3)2 as catalyst.
2 Results and discussion
The electrosynthesis of diethyl carbonate proceeds at the anode. The concomitant cathodic process could be either the reduction of PdBr2(PPh3)2 or the reduction of protons generated from EtOH. A cyclic voltammetry of a solution of PdBr2(PPh3)2 (2 mM) in ethanol (containing nBu4NBF4, 0.3 M) did not exhibit any reduction peak before the reduction of ethanol starting at –1.4 V/SCE. Consequently, the cathodic process did not involve the reduction of PdBr2(PPh3)2 to a Pd0 complex but the deprotonation of EtOH into EtO– (Eq. (3)) via the reduction of H+:
| (3) |
Cyclic voltammetry was selected as the analytical technique to monitor the fate of PdBr2(PPh3)2 in the course of the electrosynthesis. Because the catalytic precursor PdBr2(PPh3)2 could not be characterized in ethanol by cyclic voltammetry (vide supra), the mechanism of the electrosynthesis was investigated in THF to be able to monitor reactions of PdBr2(PPh3)2 with any reagents involved in the catalytic process i.e., CO and/or EtO–.
2.1 Investigation of the reaction of PdBr2(PPh3)2 with CO in the absence and presence of PPh3 (2 equiv)
The complex PdBr2(PPh3)2 (2 mM) in THF (containing nBu4NBF4, 0.3 M) was characterized by a single overall bielectronic reduction peak R1 at EpR1 = –0.875 V/SCE (gold disk electrode, i.d. 0.5 mm, scan rate: 0.2 V s–1). An oxidation peak O1 located at +0.100 V/SCE was observed on the reverse scan (Fig. 1a, full line). We have reported that the electrochemical reduction of PdBr2(PPh3)2 generates an anionic complex [Pd0(PPh3)2Br]– oxidized at O1 (Eqs. (4) and (5)) [13].
| (4) |
| (5) |
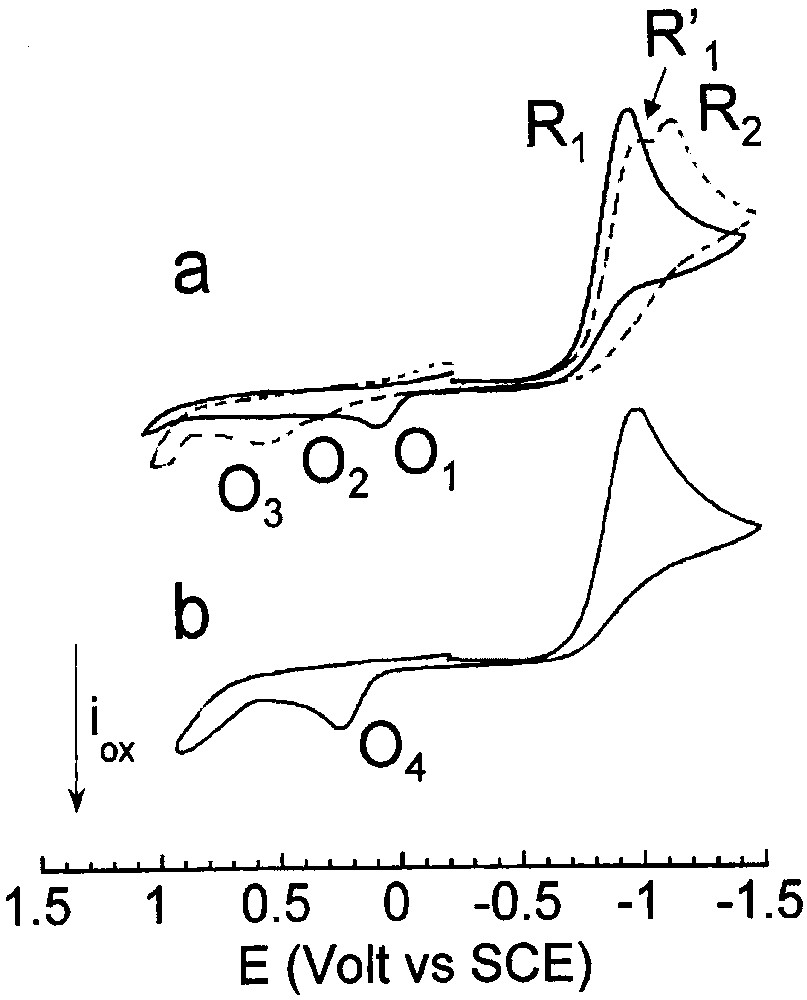
Cyclic voltammetry performed in THF (containing nBu4NBF4, 0.3 mol dm–3) at a steady gold disk electrode (i.d. 0.5 mm) with a scan rate of 0.2 V s–1 at 25 °C. Reduction first: (a) (___) PdBr2(PPh3)2 (2 mmol dm–3); (- - - ) PdBr2(PPh3)2 (2 mmol dm–3) and CO (1 atm). b) PdBr2(PPh3)2 (2 mmol dm–3), PPh3 (4 mmol dm–3) and CO (1 atm).
In the presence of excess PPh3 (2 equiv), the reduction peak R1 was not significantly modified, whereas the oxidation peak current intensity of the electrogenerated Pd0 complex located at O1 was slightly higher than in absence of PPh3, due to a stabilization of the electrogenerated Pd0 complex by PPh3 (Eq. (6)) [13].
| (6) |
Carbon monoxide was then bubbled into the cell to investigate any reaction of PdBr2(PPh3)2 with CO – complexation [12] or reduction. In the presence of CO, the reduction peak R1 of PdBr2(PPh3)2 was slightly shifted to more negative potential (EpR′1 = –0.895 V) with a lower peak current (Fig. 1a, dashed voltammogram). A new reduction peak R2 of small intensity appeared at more negative potential, EpR2 = –1.025 V (Fig. 1a, dashed voltammogram). This suggests that, in the presence of CO, bromide ions were released in higher amount in the reduction of PdBr2(PPh3)2 (Eq. (4)) than in the absence of CO. As reported, Br– ions react with PdBr2(PPh3)2 and expel one PPh3 to generate an anionic PdII complex [PdIIBr3(PPh3)]– (Eq. (7)) [14] less easily reduced at R2.
| (7) |
The potential shift observed from R1 to R′1 for PdBr2(PPh3)2 is a consequence of its involvement in the equilibrium in Eq. (7) (CE mechanism).
The complex PdBr2(PPh3)2 was thus rather stable in THF in the presence of bubbling CO, because the overall reduction current of PdBr2(PPh3)2 did not decrease with time (the current intensity of R′1 being proportional to PdBr2(PPh3)2 concentration). Consequently, at room temperature and atmospheric CO pressure, complexation of PdBr2(PPh3)2 by CO did not occur. It is known that CO can reduce PdII salts to Pd0 [15] but when ligated by phosphine, as in PdBr2(PPh3)2, the reduction by CO (under atmospheric pressure) did not occur in the time scale investigated here (1 h). The reduction of PdCl2(PPh3)2 by CO does not occur even at high CO pressure [12]. No oxidation peak was detected when the scan was performed to oxidation first.
One notices that in the presence of CO, the oxidation peak O1 of [Pd0(PPh3)2Br]–, generated by the reduction of PdBr2(PPh3)2, disappeared and two new oxidation peaks O2 (EpO2 = +0.365 V) and O3 (EpO3 = +0.595 V) were detected (Fig. 1a, dashed voltammogram). The latter was assigned to the oxidation of bromide ions by comparison to an authentic sample of nBu4NBr. This confirms that, in the presence of CO, the electrochemical reduction of PdBr2(PPh3)2 did not generate the anionic [Pd0(PPh3)2Br]– complex but a new Pd0 complex, assigned to Pd0(PPh3)2(CO) (Eq. (8)), which was oxidized at O2. However, its small oxidation peak current indicates that Pd0(PPh3)2(CO) was not very stable within the time scale of the cyclic voltammetry, due to the lack of stabilizing ligand or to insufficient CO pressure (Eq. (9), S = solvent).
| (8) |
| (9) |
The existence of the equilibrium in Eq. (9) has been evidenced by the investigation of the kinetics of oxidative addition of PhI with Pd0(PPh3)4 in the presence of CO (retarding effect of CO) [16].
In the presence of PPh3 (2 equiv) and CO, the voltammogram of PdBr2(PPh3)2 exhibited the only reduction peak R1 (Fig. 1b), because the equilibrium in Eq. (7) was shifted towards its left hand-side due to the excess PPh3. On the reverse scan, a single oxidation peak was detected at O4 (EpO4 = +0.245 V), whose peak current was much higher than that of O2 (compare Fig. 1a and b).
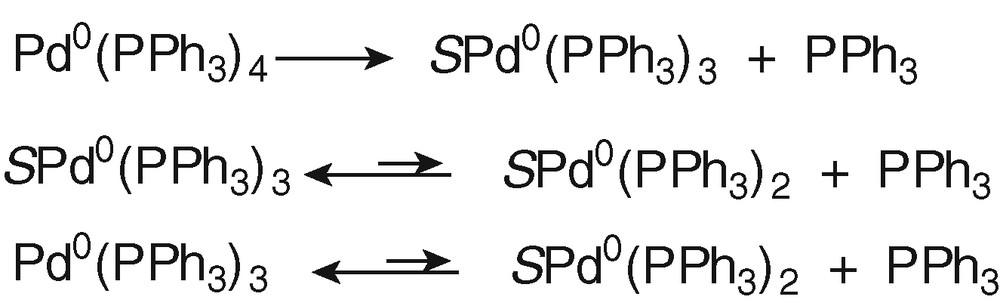
To characterize the Pd0 complex that was oxidized at O4, a set of experiments was performed with an authentic sample of Pd0(PPh3)4, which contains 4 equiv PPh3 per Pd0 as when PdBr2(PPh3)2 was reduced in the presence of 2 equiv PPh3. In THF, Pd0(PPh3)4 dissociates to Pd0(PPh3)3, SPd0(PPh3)3 and SPd0(PPh3)2 (Scheme 1, S = solvent) [17].
This is why the voltammogram of Pd0(PPh3)4 (2 mM) in THF (containing nBu4NBF4, 0.3 M) exhibited two close oxidation peaks O′1 (EpO′1 = +0.245 V) and O'2 (EpO′2 = +0.375 V) (Fig. 2a) assigned to Pd0(PPh3)3 and SPd0(PPh3)3 involved in the equilibrium of Scheme 1 [17]. When Br– ions (2 equiv) were added to Pd0(PPh3)4, the two oxidation peaks O′1 and O'2 disappeared to give rise to a single oxidation peak O1 (Fig. 2b), already obtained when PdBr2(PPh3)2 was reduced in the presence of 2 equiv PPh3 and which characterized the oxidation of the anionic Pd0 complexes [Pd0(PPh3)nBr]– (n = 2 and 3) involved in the equilibrium in Eq. (6). When CO was bubbled into the solution of Pd0(PPh3)4 (2 mM) in THF containing 2 equiv Br–, a new broad oxidation peak developed at O4 (EpO4 = +0.245 V) (Fig. 2c), attesting the complexation of the [Pd0(PPh3)nBr]– complexes by CO to form less easily oxidized neutral complexes Pd0(PPh3)n(CO) (n = 2 and 3) (Eq. (10)) – for the existence and characterization of Pd0(PPh3)n(CO) complexes, see [18]. The oxidation peak O4 is similar to that observed when PdBr2(PPh3)2 was reduced in the presence of CO and 2 equiv PPh3 (Fig. 1b).
| (10) |
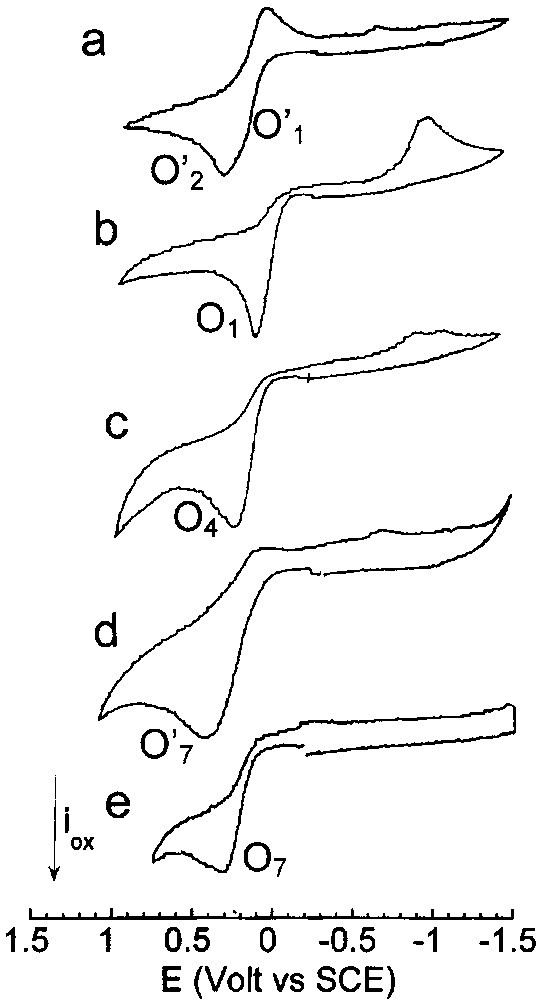
Cyclic voltammetry performed in THF (containing nBu4NBF4, 0.3 mol dm–3) at a steady gold disk electrode (i.d. 0.5 mm) with a scan rate of 0.2 V s–1 at 25 °C. Oxidation first: (a) Pd0(PPh3)4 (2 mmol dm–3), (b) Pd0(PPh3)4 (2 mmol dm–3) and nBu4NBr (4 mmol dm–3), (c) Pd0(PPh3)4 (2 mmol dm–3), nBu4NBr (4 mmol dm–3) and CO (1 atm), (d) Pd0(PPh3)4 (2 mmol dm–3), EtONa (16 mmol dm–3) and CO (1 atm), (e) Pd0(PPh3)4 (2 mmol dm–3), nBu4NBr (4 mmol dm–3) EtONa (16 mmol dm–3) and CO (1 atm).
In another set of experiments, CO was first added to Pd0(PPh3)4. A broad oxidation peak O′4 was observed at EpO′4 = +0.485 V which characterized the oxidation of the Pd0(PPh3)n(CO) (n = 2 and 3) complexes involved in the equilibrium of Eq.(10). After addition of 2 equiv Br–, the broad oxidation peak O′4 disappeared and the only peak O4 (EpO′4=+0.241V) was detected. Since we have seen above that Br– was released from anionic [Pd0(PPh3)2Br]– in the presence of CO, the peak O4 characterized the oxidation of Pd0(PPh3)n(CO) (n = 2 and 3) in the presence of Br–. The observed potential shift from O′4 to O4 was therefore mostly due to the chemical reaction of Br– with the transient PdI electrogenerated in the first electron oxidation of Pd0(PPh3)n(CO) (n = 2, 3) (EC mechanism). The potential of an irreversible peak depends on the rate of the chemical step that follows the first electron transfer. In the present case, the first electron transfer gives a cationic PdI complex involved in a chemical reaction with Br– ions. This transient ‘PdIBr’ complex is more easily oxidized than the initial Pd0. This results in an overall bielectronic oxidation process occurring at the oxidation potential of Pd0, which gives eventually a PdII complex. For the mechanism of the electrochemical oxidation of Pd0 complexes, see [19]. The above results are summarized in Scheme 2.
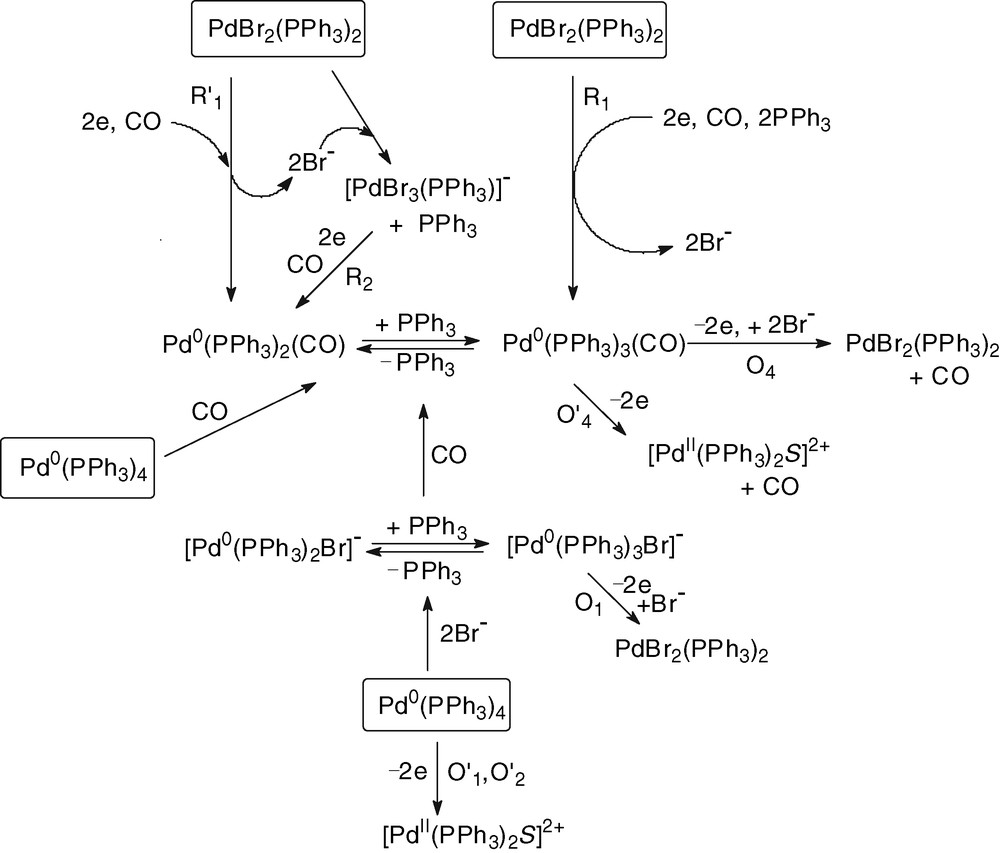
Whatever the Pd0 precursor, Pd0(PPh3)4 and 2 equiv Br– or the Pd0 complex generated by the electrochemical reduction of PdBr2(PPh3)2 in the presence of 2 equiv PPh3, the introduction of CO generates Pd0(PPh3)n(CO) (n = 2 and 3) complexes which are oxidized at O4 (EpO4 = +0.245 V, Figs. 1b and 2c).
At room temperature and under atmospheric pressure, CO does not coordinate PdBr2(PPh3)2 and does not reduce PdBr2(PPh3)2. The only evolution of PdBr2(PPh3)2 would then be its reduction by the anions ethoxide generated from EtOH in the cathodic process (Eq. (3)).
2.2 Reduction of PdBr2(PPh3)2 by ethoxide ions in the absence and presence of PPh3 (2 equiv)
The fate of PdBr2(PPh3)2 in the presence of excess EtONa (used to mimic the effect of EtO– electrogenerated at the cathode in the catalytic reaction) was monitored by cyclic voltammetry in THF. The oxidation potential of EtO– was observed at +1.175 V in THF.
When 8 equiv EtO– were added to a solution of PdBr2(PPh3)2 (2 mM) in THF, its reduction peak R1 disappeared and a succession of oxidation peaks were observed (Fig. 3), whereas the solution turned black. This suggests that PdBr2(PPh3)2 was reduced by the ethoxide to a multitude of Pd0 complexes or clusters due to a lack of stabilizing ligands.
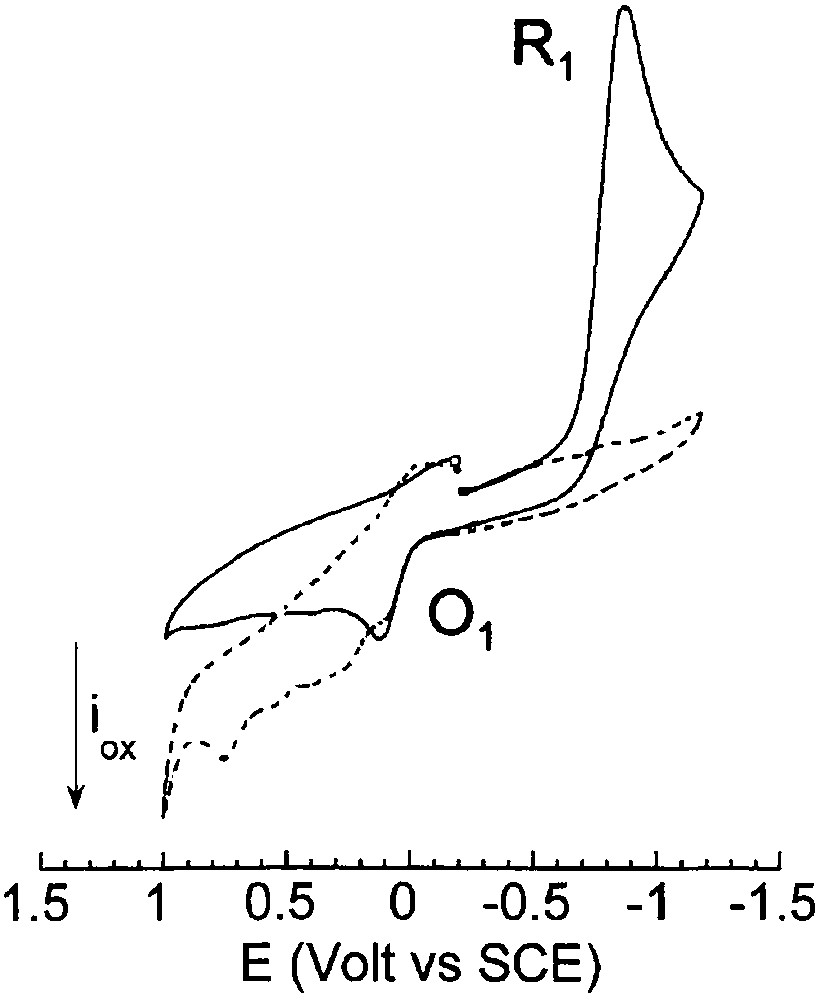
Cyclic voltammetry performed in THF (containing nBu4NBF4, 0.3 mol dm–3) at a steady gold disk electrode (i.d. 0.5 mm) with a scan rate of 0.2 V s–1 at 25 °C. Reduction first: (___) PdBr2(PPh3)2 (2 mmol dm–3); (- - - ) PdBr2(PPh3)2 (2 mmol dm–3) and EtONa (16 mmol dm–3).
To observe stable Pd0 complex(es) detectable in cyclic voltammetry, the chemical reduction of PdBr2(PPh3)2 by the ethoxide was performed in the presence of two equiv PPh3. The reduction of PdBr2(PPh3)2 by 2 equiv EtO– was a slow reaction as attested by the slow decay of the reduction peak of PdBr2(PPh3)2. A large excess EtO– (8 equiv) was required to reduce PdBr2(PPh3)2 quantitatively within a reasonable time (1 h 30). Fig. 4a exhibits the reduction peak of PdBr2(PPh3)2 in the presence of 2 equiv PPh3. When 8 equiv EtO– were added, the reduction peak current of PdBr2(PPh3)2 decreased (Fig. 4b, reaction time: 35 min) attesting a reaction of PdBr2(PPh3)2 with EtO–. An oxidation peak was observed on the reverse scan at a potential close to that of O1, but with a higher current intensity than initially (compare Fig. 4a and b), attesting that a Pd0 complex was generated by the reaction of EtO– with PdBr2(PPh3)2. This Pd0 complex was indeed detected by its oxidation peak O5 when the voltammetry was performed directly towards positive potentials, without scanning over peak R1 (Fig. 4c). The complete reduction of PdBr2(PPh3)2 by EtO– was achieved within 1 h and a half and the resulting Pd0 was oxidized at O5 (EpO5 = +0.090 V).
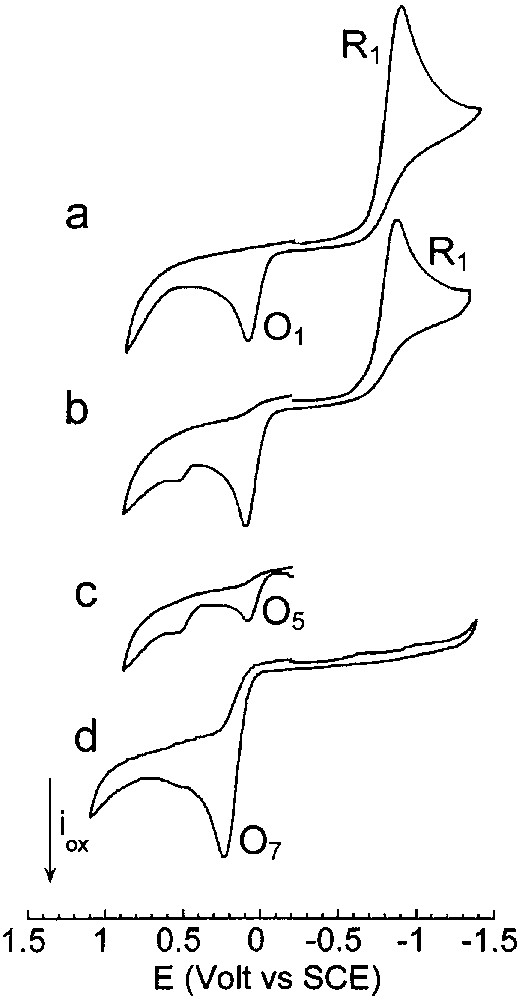
Cyclic voltammetry performed in THF (containing nBu4NBF4, 0.3 mol dm–3) at a steady gold disk electrode (i.d. 0.5 mm) with a scan rate of 0.2 V s–1 at 25 °C. Reduction first: (a) PdBr2(PPh3)2 (2 mmol dm–3) and PPh3 (4 mmol dm–3); (b) PdBr2(PPh3)2 (2 mmol dm–3), PPh3 (4 mmol dm–3) and EtONa (16 mmol dm–3), 35 min after addition of EtONa; (c) oxidation first: PdBr2(PPh3)2 (2 mmol dm–3), PPh3 (4 mmol dm–3) and EtONa (16 mmol dm–3), 40 min after addition of EtONa; (d) reduction first: PdBr2(PPh3)2 (2 mmol dm–3), PPh3 (4 mmol dm–3), EtONa (16 mmol dm–3) and CO (1 atm) after 1 h 30. Masquer
Cyclic voltammetry performed in THF (containing nBu4NBF4, 0.3 mol dm–3) at a steady gold disk electrode (i.d. 0.5 mm) with a scan rate of 0.2 V s–1 at 25 °C. Reduction first: (a) PdBr2(PPh3)2 (2 mmol dm–3) and PPh3 (4 mmol dm–3); (b) PdBr2(PPh3)2 (2 mmol ... Lire la suite
Due to the close vicinity of the oxidation potentials of O1 and O5, it is difficult to conclude from the cyclic voltammetry whether the Pd0 generated by the chemical reduction of PdBr2(PPh3)2 was still ligated by Br– or by EtO–. Alcazar-Roman and Hartwig have reported kinetic evidence of the formation of anionic Pd0 complexes ligated by alkoxides [20]. To better characterize the Pd0 complex generated by the chemical reduction, EtO– (8 equiv) was added to a solution of Pd0(PPh3)4 (2 mM) in THF. The two oxidation peaks O′1 (+0.245 V) and O’2 (+0.375 V) characteristic of Pd0(PPh3)3 and SPd0(PPh3)3 [17] were slightly shifted to more positive potentials O′5 (+0.265 V) and O′6 (+0.385 V) respectively. One would have expected to observe less positive oxidation potentials for the putative anionic [Pd0(PPh3)n(OEt)]– species. However, the electrochemical oxidation of anionic [Pd0(PPh3)n(OEt)]– species in the presence of EtO– must be a complex process as attested by the irreversibility of the oxidation peaks. The formation of anionic [Pd0(PPh3)n(OEt)]– (n = 2 and 3) complexes was better supported by 31P NMR experiments. Indeed, the broad 31P NMR signal at +3.23 ppm characteristic of Pd0(PPh3)n (n = 2 and 3) in equilibrium with PPh3, generated from Pd0(PPh3)4 (7.9 mM) in THF (Scheme 1), was shifted to –3.0 ppm by addition of EtO– (8 equiv). The signal of the free phosphine was not observed. This shows that EtO– interfered in the equilibrium of Scheme 1, to form anionic species (Eq. (11)), as already established for halides or acetates [21].
| (11) |
The chemical reduction of PdBr2(PPh3)2 requires two equivalents of EtO– and the mechanism involves a classical β–hydride elimination [22], leading to a hydrido-PdII complex, whose HBr elimination by the basic ethoxide generates a Pd0 complex stabilized by excess EtO– and PPh3 (Scheme 3).
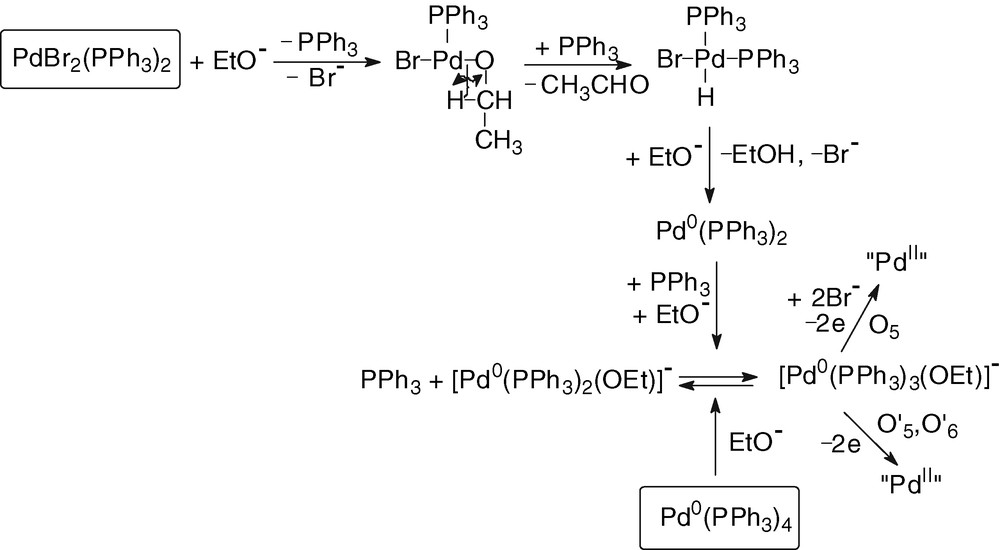
2.3 Reaction of ethoxide ions with Pd0(PPh3)n(CO) complexes
According to Scheme 2, Pd0(PPh3)n(CO) (n = 2 and 3) complexes were generated in the absence of EtO–, whereas [Pd0(PPh3)n(OEt)]– complexes were formed in the absence of CO (Scheme 3). Taking into account the fact that (i) CO is a better ligand than EtO– for the electron rich Pd0(PPh3)2 complex and that (ii) CO is present in large amount in the very first step of the electrolysis whereas EtO– is generated only when the electrolysis proceeds (Eq. (3)), we favour the formation of Pd0(PPh3)2(CO) as the main Pd0 complex in the very first step of the electrolysis (performed in the absence of PPh3) rather than that of [Pd0(PPh3)2(OEt)]–. It was thus of interest to investigate the reaction of EtO– with Pd0(PPh3)n(CO) complexes, which was monitored in parallel by 31P NMR spectroscopy and cyclic voltammetry.
Pd0(PPh3)n(CO) (n = 2 and 3) complexes were first generated by bubbling CO into a solution of Pd0(PPh3)4 (8 mM) in THF and characterized by a broad 31P NMR signal at +7.43 ppm (Eq. (10)) and the broad oxidation peak O′4 (EpO′4 = +0.485 V) already mentioned above. Addition of EtO– (8 equiv) generated a new broad signal at +4.15 ppm and a new oxidation peak at less positive potential O′7 (EpO′7 = +0.415 V) (Fig. 2d), i.e., new species. The reaction of strong nucleophiles such as alkoxydes RO– with M0(CO)n complexes is a known process that involves the nucleophilic attack of one CO ligand to form anionic alkoxycarbonyl [(CO)n–1M0(COOR)]– complexes [23,24]. In the present case, anionic Pd0 complexes [(PPh3)nPd0–COOEt]– (n = 2 and 3) would be generated (Scheme 4). The reaction was monitored by IR spectroscopy. The CO vibration in Pd0(PPh3)3(CO) appeared at 2019 cm–1 in THF, close to that reported for the solid (1955 cm–1) [18]. When EtO– was added to Pd0(PPh3)3(CO), a new absorption developed at 1650 cm–1, characteristic of COOEt ligated to a palladium centre [11].
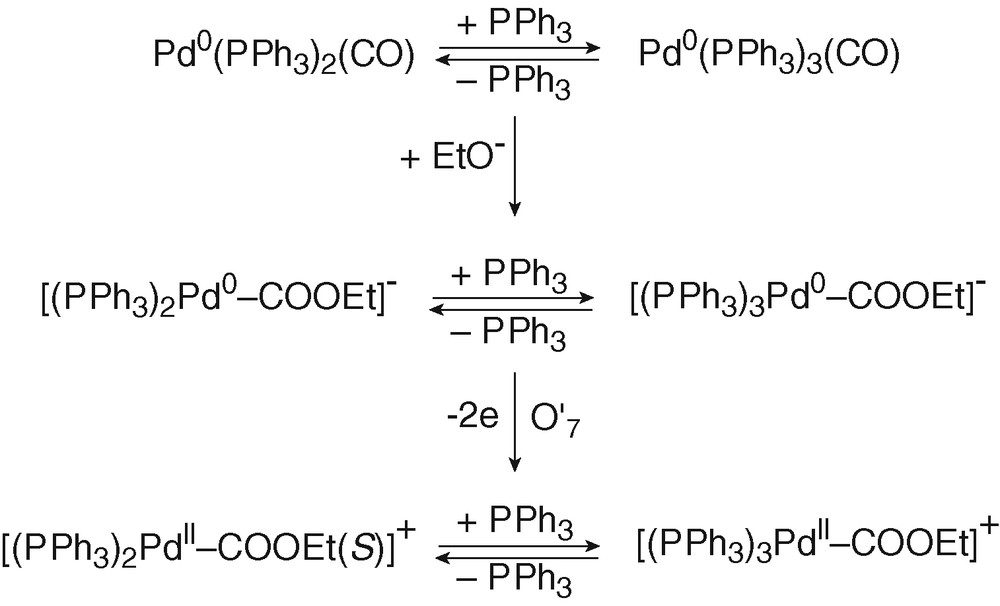
The bielectronic oxidation of [(PPh3)nPd0–COOEt]– (n = 2 and 3) at O′7 is expected to generate cationic PdII complexes [(PPh3)nPdII–COOEt]+ (n = 2 and 3) (Scheme 4).
When 2 equiv of Br– were added to the solution containing of [(PPh3)nPd0–COOEt]– (n = 2 and 3), the oxidation peak O'7 (EpO′7=+0.415V) observed in Fig. 2d was replaced by a new one O7 located at less positive potential (EpO7 = +0.215 V) (Fig. 2e). The easier oxidation of [(PPh3)nPd0–COOEt]– (n = 2 and 3) in the presence of Br– is most certainly featuring the chemical reaction of the bromide ion with the transient PdI formed in the first electron transfer (EC mechanism). After an overall bielectronic process, a neutral PdII complex BrPdII–COOEt(PPh3)2 should be generated ultimately (Scheme 5). Related complexes ClPd–COOR(PPh3)2 are reported in literature. They have been synthesized by either the oxidative addition of Cl–COOR to Pd0(PPh3)4 [11,25,26].
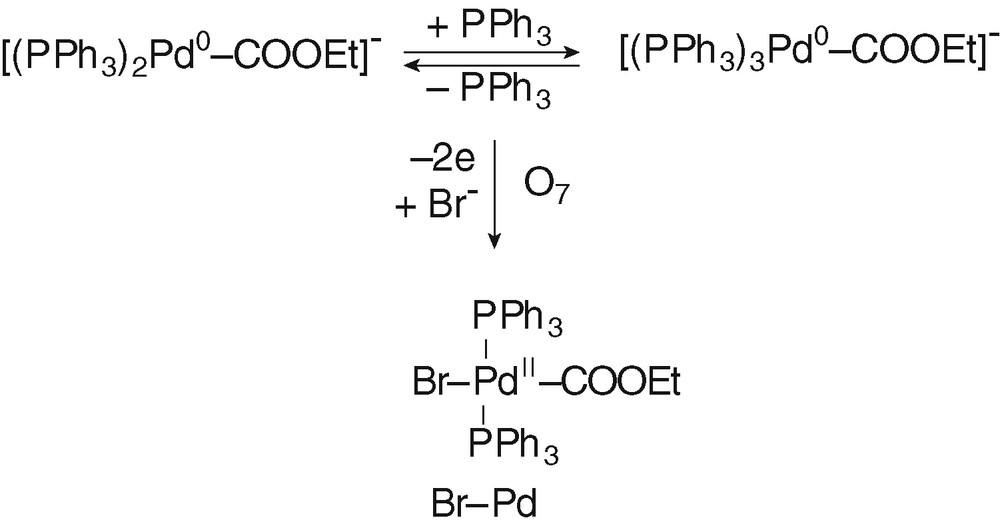
The same oxidation peak O7 was also observed when PdBr2(PPh3)2 was reduced by EtO– in the presence of PPh3 (2 equiv) and CO (Fig. 4d).
2.4 Mechanism of the palladium-catalysed electrosynthesis of diethyl carbonate from carbon monoxide and ethanol
The above mechanistic investigations were developed in THF and in the presence of excess PPh3, so that to have a chance to observe and characterize relatively stable Pd0 complexes and to be able to monitor their evolution in the presence of the reagents involved in the catalytic process. The electrosynthesis of diethyl carbonate proceeds in ethanol and in the absence of extra PPh3 [3]. The catalyst PdBr2(PPh3)2 was not tested in the electrosynthesis of diethyl carbonate but a related complex PdCl2(PPh3)2. The latter was also successfully used in the electrocarbonylation of benzylamine to dibenzylurea [27]. A catalytic cycle for the electrosynthesis performed in ethanol is proposed in Scheme 6.
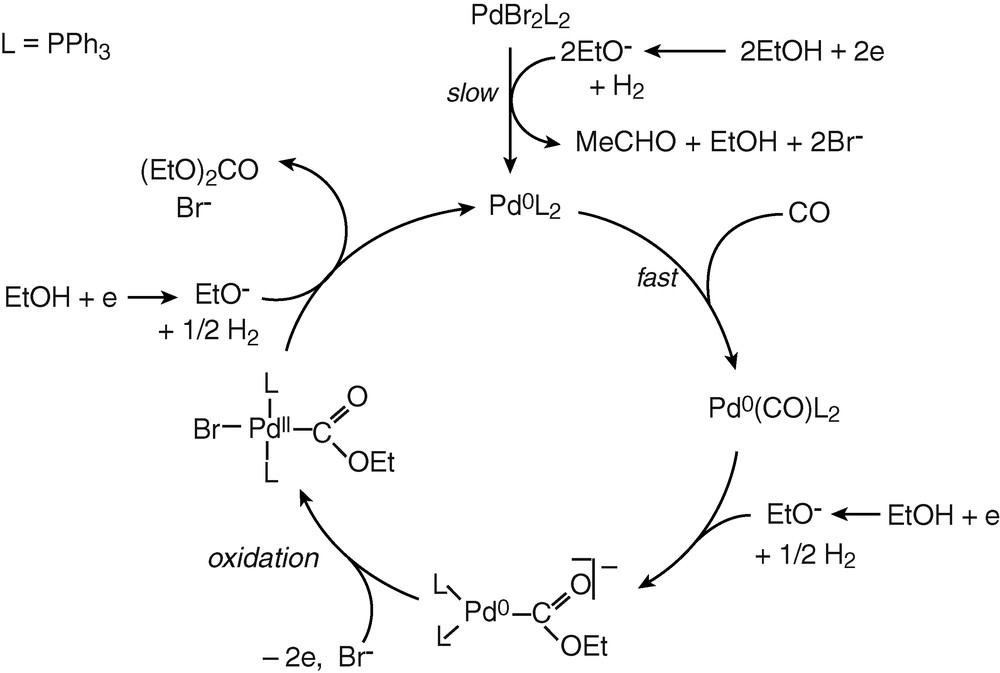
The chemical reduction of the catalytic precursor PdBr2(PPh3)2 by the EtO– generated in the cathodic process gives a Pd0 that is probably not ligated by neither Br– nor EtO–, but by CO. Indeed, the slow chemical reduction proceeds in the presence of CO and, as mentioned above, the concentration of CO in the electrolysis is constant (bubbling CO) and is always higher than that of EtO– anions. The latter are generated at the cathode and their concentration is controlled by the current density as well as by their chemical consumption. Indeed, once EtO– ions have reduced the PdBr2(PPh3)2 to Pd0, they are also consumed in a chemical reaction with Pd0(PPh3)2(CO). This reaction gives the anionic complex [(PPh3)2Pd0–COOEt]–. The key step of the anodic process is thus the overall bielectronic oxidation of [(PPh3)2Pd0–COOEt]–. This oxidation is conducted in the presence of Br– delivered by the precursor PdBr2(PPh3)2. Consequently, it should generate the neutral complex BrPdII–COOEt(PPh3)2 rather than the cationic complex [(PPh3)2PdII–COOEt(S)]+.
The last step of the catalytic cycle is a nucleophilic attack of the COOEt ligand of BrPdII–COOEt(PPh3)2 by EtO–, as detailed in Scheme 7. This reaction is supported by reported nucleophilic attack of methanol on related complexes, (AcO)PdII–COOMe(PPh3)2, which yields dimethyl carbonate only in the presence of a base [11].
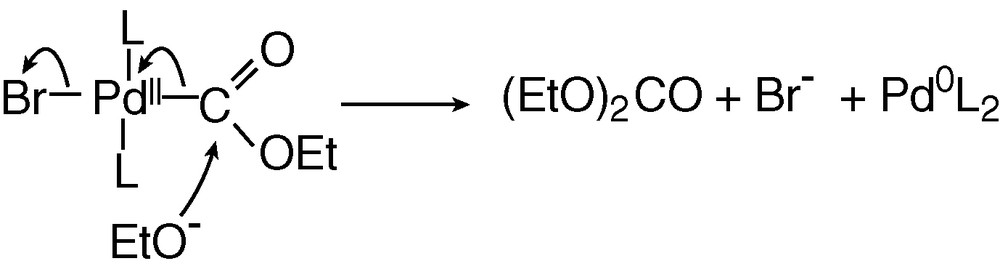
The mechanism of the palladium-catalysed electrosynthesis of diethyl carbonate from carbon monoxide and ethanol described in Scheme 6 indicates that the anodic and cathodic reactions are interfering. As a result of the mechanistic investigation, the two processes may be expressed as:
In the presence of bromide ions, which occurs in the electrolysis because of the catalytic precursor PdBr2(PPh3)2, the oxidation potential of Pd0(PPh3)2(CO) (EpO4 = +0.245 V) and that of [(PPh3)2Pd0–COOEt]– (EpO7 = +0.215 V) are very close. If the nucleophilic attack of Pd0(PPh3)2(CO) by EtO– is not fast enough (it is limited by the rate of production of EtO– at the cathode) the oxidation of Pd0(PPh3)2(CO) will also occur leading back to PdBr2(PPh3)2 and consequently would contribute to lower the faradic efficiency.
3 Conclusion
A mechanism is proposed for the electrosynthesis of diethyl carbonate from carbon monoxide and ethanol catalysed by PdBr2(PPh3)2, proceeding at room temperature and under atmospheric CO pressure (Scheme 6). The cathodic and anodic processes have been defined. PdBr2(PPh3)2 does not react with CO but is reduced by EtO–. The catalytic cycle involves a succession of neutral or anionic Pd0 and PdII complexes generated in chemical steps or in electrochemical oxidation steps.
4 Experimental
The 31P NMR spectra were recorded on a Bruker spectrometer (162 MHz) using H3PO4 as an external reference. IR spectra were recorded on a Nicolet Impact 400D spectrometer. Cyclic voltammetry was performed with a homemade potentiostat and a waveform generator GSTP4 (Radiometer Analytical). The voltammograms were recorded with an oscilloscope Nicolet 301.
All experiments were performed using standard Schlenk techniques under argon atmosphere. THF was distilled on potassium/benzophenone. PPh3, anhydrous nBu4NBr, and ethanol, sodium ethoxide were commercial (Aldrich) and used without purification. PdBr2(PPh3)2 [13] and Pd0(PPh3)4 [28] was synthesized according to published procedures.
4.1 Electrochemical set-up for cyclic voltammetry
Electrochemical experiments were carried out in a three-electrode cell connected to a Schlenk line. The cell was thermostated at 25 °C. The steady working electrode consisted of a gold disk (i.d. 0.5 mm). The counter electrode was a platinum wire of ca 1-cm2 apparent surface area. The reference was a saturated calomel electrode separated from the solution by a bridge filled with a solution of nBu4NBF4 (0.3 mol dm–3) in 3 ml of THF. 12 ml of THF containing the same concentration of supporting electrolyte was poured into the cell followed by 17 mg (0.024 mmol) of PdBr2(PPh3)2 and 12.5 mg (0.048 mmol) of PPh3, when required.
In other experiments, 13 mg (0.19 mmol) of EtONa were added to the solution of PdBr2(PPh3)2 in the absence or presence of bubbling CO.
The cyclic voltammetry was also performed on solutions of 28 mg (0.024 mmol) Pd0(PPh3)4 alone and then in the presence of 13 mg (0.19 mmol) of EtONa in the absence or presence of bubbling CO. When required, 15.5 mg (0.048 mmol) of nBu4NBr were added.
Acknowledgements
This work has been supported in part by the ‘Centre national de la recherche scientifique’’ (CNRS, UMR 8640 ‘PASTEUR’), the ‘Ministère de la Recherche (‘École normale supérieure’) and the University Paris-6. We thank Johnson Matthey for a generous loan of sodium tetrachloropalladate.
Vous devez vous connecter pour continuer.
S'authentifier