

1 Introduction
Ever since the parallel discovery by the groups of Brookhart [1] and Gibson [2] of the performance of iron and cobalt complexes bearing pyridine diimine ligands in the field of olefin oligomerisation and polymerisation, numerous studies have been dedicated to this particular ligand [3]. In the case of cobalt, we [4] and others [5] showed that, in the activation pathway, MAO reduces the dichloride complex prior to alkylation. Several other alkylating agents could achieve this as well.
Complexes of other metals show different behaviour in reaction with alkylating agents [6–12]. Of particular interest to the present work is the reaction of LCrCl3 [L = 2,6-bis(1-(2,6-diisopropylphenylimino)ethyl)pyridine] with three equivalents of BzMgCl to form an unexpected dimer (Fig. 1) [10]. This was suggested to involve reduction to LCrCl2, alkylation of the metal centre and the pyridine C4 position to give [L+Bz]CrBz and finally dimerisation via a double carbon–carbon coupling at the pyridine C3,5 positions (see Fig. 1).
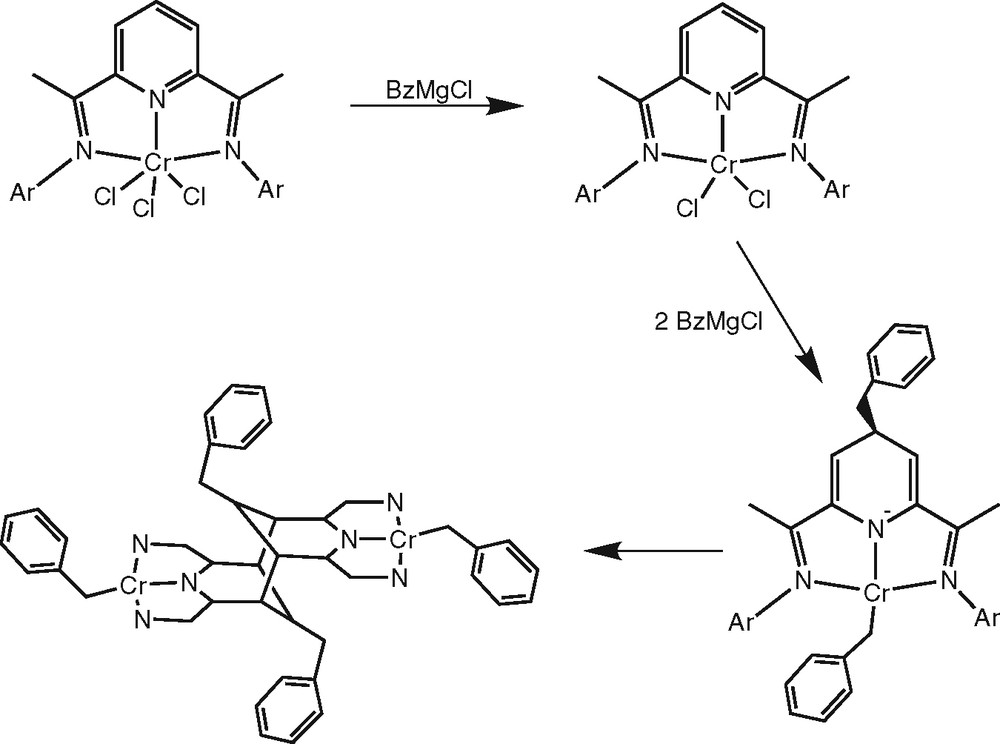
Reaction mechanism for the double C–C coupling in the chromium complex as proposed by Gambarotta et al. [10].
We have been studying the reaction of L with alkylaluminum compounds and found that it is unexpectedly complex and does not always only yield the imine adducts described by Gibson and Grassi [11,12]. In the present preliminary communication, we report, as a part of this study, the formation of a tricyclic dimer similar to the one described by Gambarotta [10], but formed in the absence of any transition metal.
2 Results and discussion
2.1 General
2.1.1 L + AlEt2Cl
Immediately upon mixing L and one equivalent of AlEt2Cl in toluene, a colour change from yellow to green is observed. Refluxing (110 °C) this solution overnight resulted in a colour change to deep purple. 1H-NMR showed that a complex mixture of products has formed. By crystallisation, we were able to structurally identify two components out of this mixture. According to 1H and 13C NMR, the mixture contains several additional components, and clear separation proved impossible. Also, we cannot assign the NMR signals of 1 in the mixture, so we cannot estimate how much of it is actually formed in this reaction. Details on the reaction of several alkylaluminum compounds will be reported in a forthcoming full paper (Q. Knijnenburg, T.Vissers, P.H.M. Budzelaar, to be published). From the purple solution in toluene, very dark purple trapezium-shaped crystals of 1 could be grown at room temperature. On cooling the mother liquor to –40°C, a few cube-like light-orange crystals were formed as well. The orange crystals were of very poor quality. X-ray diffraction studies, though inconclusive, indicated the presence of an imine adduct 5 (see supporting information) similar to the ones described by the groups of Gibson and Milione (Scheme 1) [11,12].
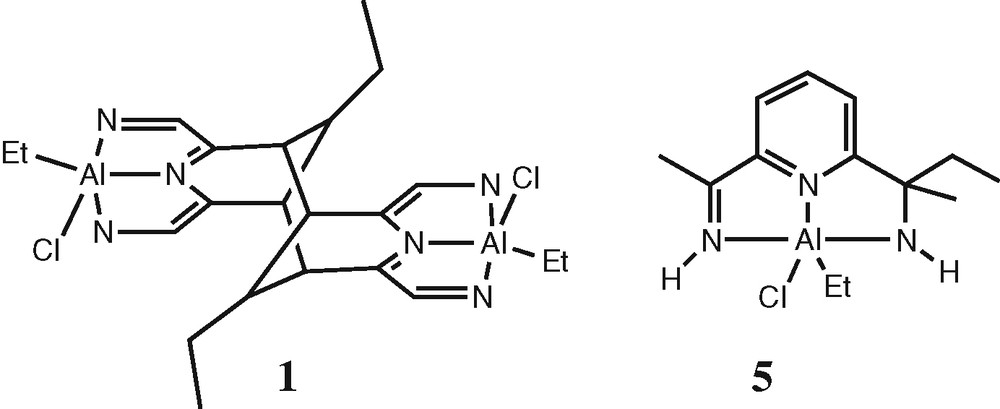
Structures of 1 and 5.
The purple crystals proved to contain a dinuclear species, resulting from coupling of two separate molecules at the C3 and C5 positions, presumably preceded by an attack of an ethyl group at the C4 position (Fig. 2). It is very similar to the dimer described by Gambarotta et al. for chromium [10], as mentioned in the introduction. Remarkably, no transition metal seems to be necessary for the generation of this unusual tricyclic ligand skeleton. We will return to the mechanism of its formation later on.
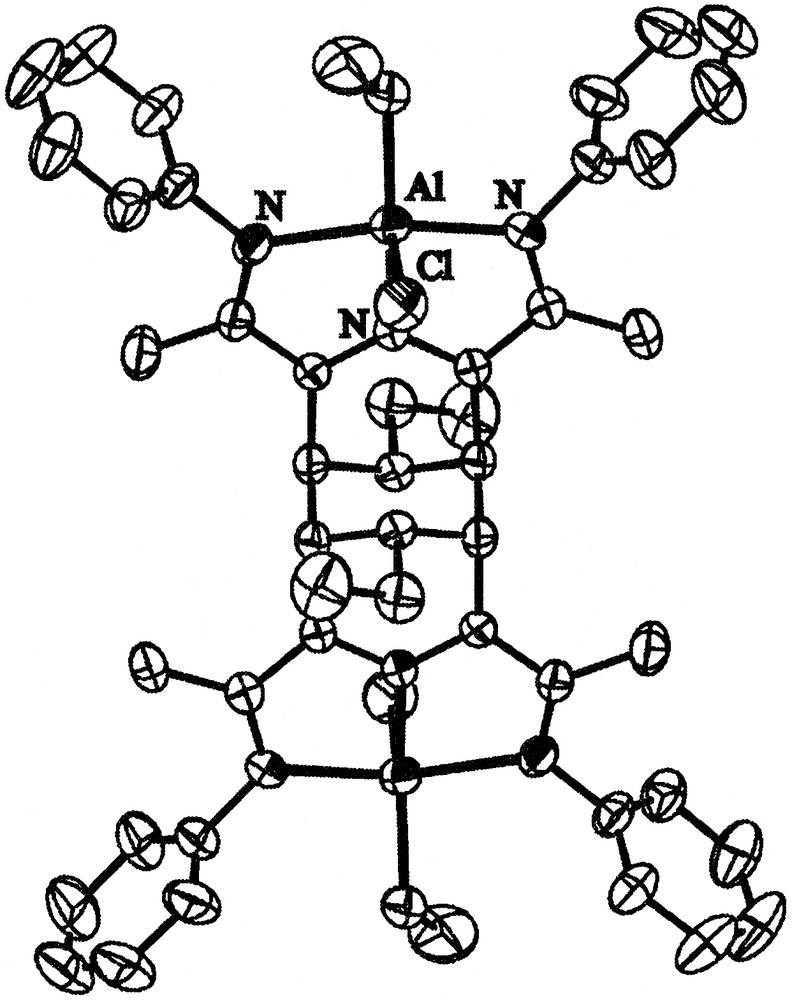
Thermal ellipsoid of 1, drawn at 50% probability. Hydrogens and isopropyl groups are omitted for reasons of clarity.
The geometry around the aluminium centres is distorted square pyramidal. The distances of 1 are shown in Fig. 3, together with those of the calculated structure 1a (vide infra) and of the similar chromium compound 2 described by Gambarotta [10]. The distances suggest a conjugated π system for the NCCNCCN framework.
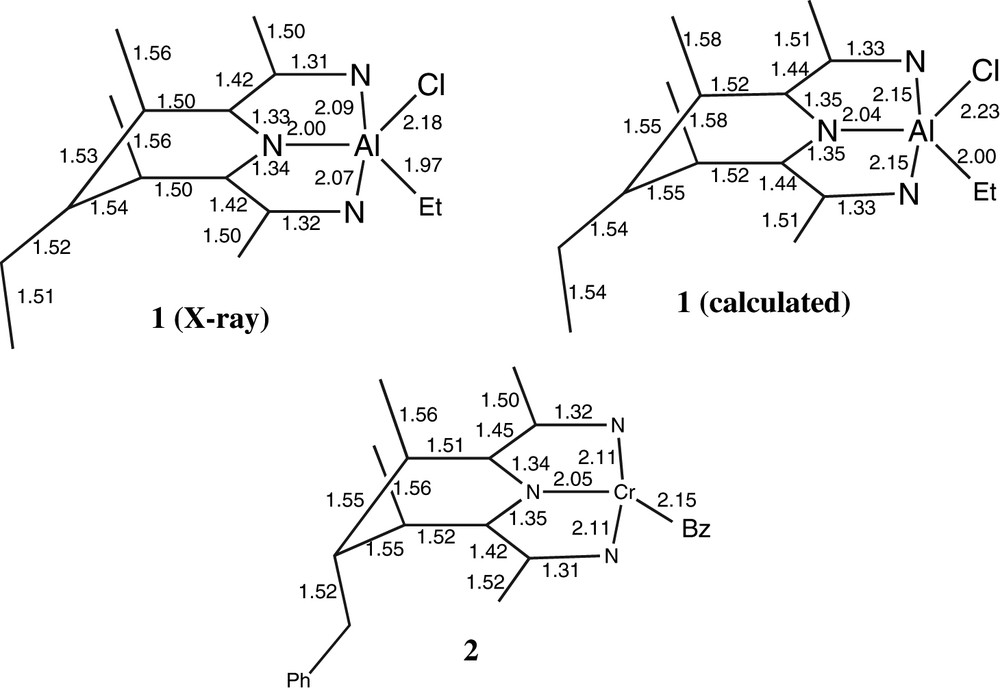
Bond distances (Å) of the structure of 1 (X-ray and calculated) and of the chromium dimer 2 [10].
The calculated structure for model complex 1a lacking the bulky aryl groups (vide infra) has a trigonal bipyramidal geometry. Distortions from trigonal bipyramidal to square pyramidal as seen in 1, induced by the bulky 2,6-iPrC6H3 substituents, have been observed before [13,14]. The conjugated frameworks are very similar in the three structures; the main difference between 1 and 2 is the somewhat larger size of the metal in 2.
2.2 Computational studies
Dimer formation was studied by unrestricted DFT methods. As a model system we used the reaction between simplified ligand L′ and AlEt2Cl (Fig. 4).
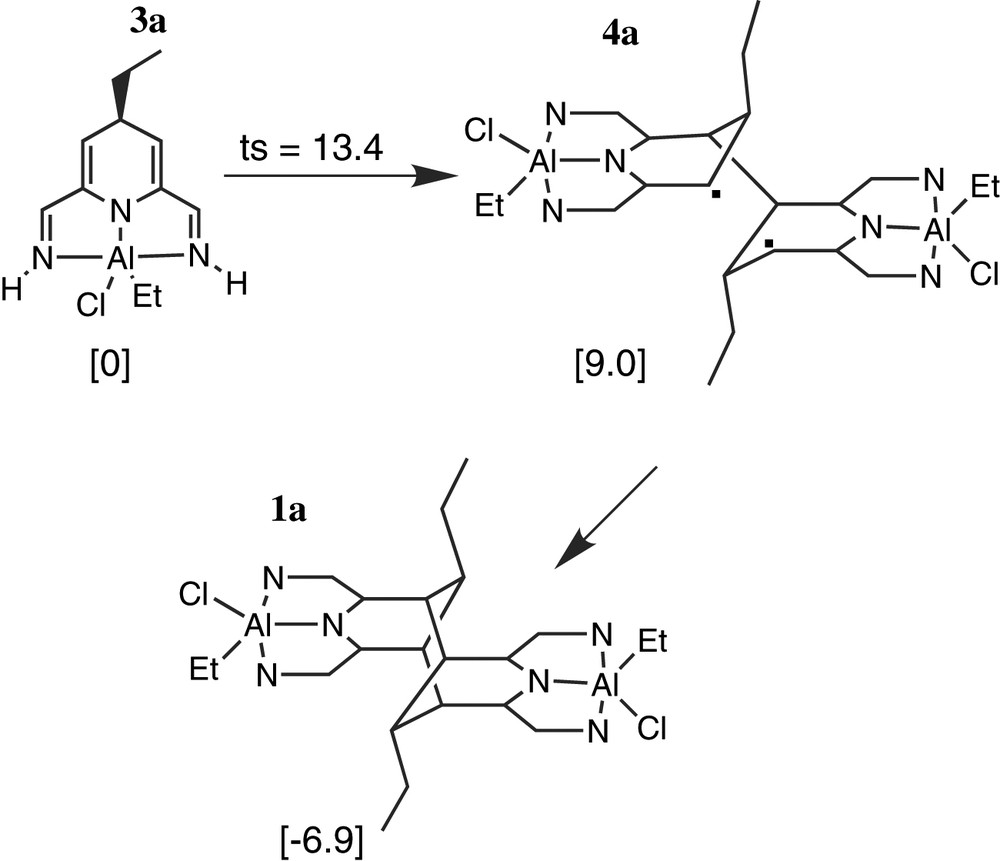
Reactions studied computationally, with energies relative to monomer 3a of minima and transition states in kcal mol–1.
Approach of two monomers of C4-adduct 3a easily leads to single C–C coupling with a modest barrier of 13.4 kcal mol–1. Product 4a is essentially a biradical (<S2> = 1.01). After coupling, the two halves of the molecule rotate with respect to each other around the new C–C bond to let the radical centres approach each other and form the second C–C bond. The energy profile for this rotation is extremely flat. The final stages of the approach were followed by constrained geometry optimisations; Fig. 5 shows the energy profile and the decrease in biradical character on giving final product 1a.
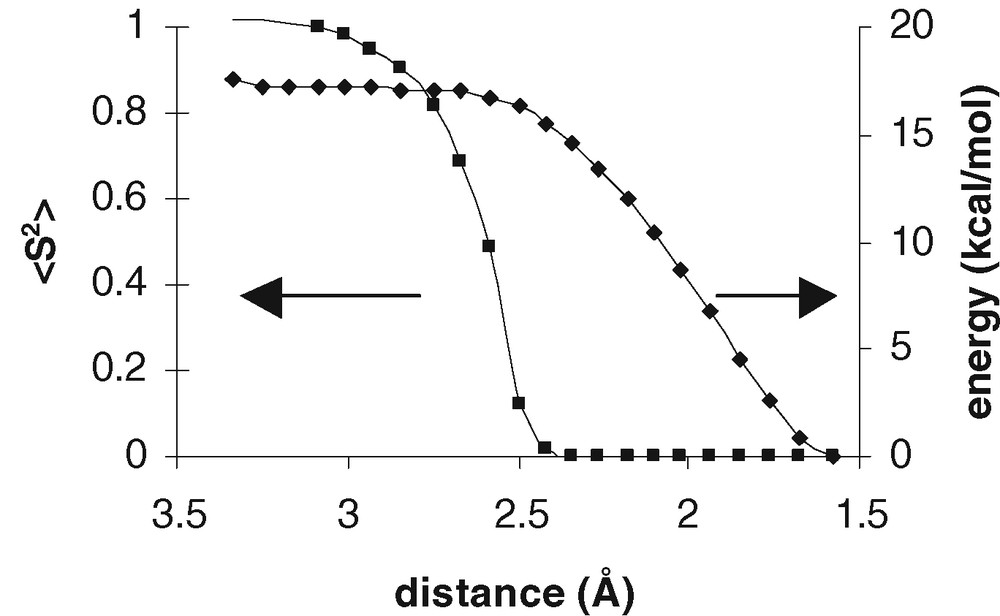
Profile for formation of the second C–C bond of 1a: energy relative to dimer 1a (●) and <S2> value (■)
The direct, synchronous 2+2 dimerisation path from 3a to 1a is symmetry-forbidden (alternative, synchronous and closed-shell approaches were examined but did not lead to low-energy dimerisation paths). This explains why, in our calculations, a non-synchronous biradical path is followed. The barrier for this path is surprisingly low. We believe that this is caused by the more efficient delocalisation of negative charge over the two imine nitrogens in 4a and 1a (in 3a, it is formally localized on the pyridyl nitrogen). In addition, 3a is cross-conjugated, which is in general less favourable than the linear conjugation over the NCCNCCN path seen in 1a. However, we do not believe that the calculated barrier of 13.4 kcal mol–1 for the biradical path is very accurate, since it is notoriously difficult to calculate accurate relative energies for open-shell and closed-shell species. Presumably, the most one can say on the basis of our calculations is that C–C coupling is ‘relatively easy’.
3 Conclusion
Our work shows that in the reaction between L and AlEt2Cl a tricyclic dimer is formed, which is very similar to the one reported earlier for chromium. However, in our case, the dimerisation was accomplished in the absence of a transition metal. DFT calculations show that the dimerisation follows a non-synchronous biradical pathway. The newly formed six-membered ring has all substituents in defined stereoselective orientation, which suggests that this reaction might have some potential in organic synthesis.
4 Experimental section
4.1 General
All manipulations were carried out under an atmosphere of argon using standard Schlenk techniques or in a conventional nitrogen-filled glovebox. Toluene was distilled over Na/benzophenone prior to use. NMR spectra were recorded on Bruker spectrometers at ambient temperature. All chemicals were obtained commercially and used as received. Ligand L was synthesized according to literature procedures [13].
4.1.1 L + AlEt2Cl
508 mg L (1.05 mmol) and 1.15 ml 1 M AlEt2Cl (1.15 mmol; 1.08 equiv) were put in 30 ml toluene. The green solution was refluxed overnight, which made the colour change to purple. Upon storage of this purple solution, a small amount of very small, dark purple, trapezium-shaped crystals were formed that were characterized by X-ray difffraction as dimer 1. Cooling of the mother liquor offered a few cubic orange crystals of 5, also characterized by X-ray difffraction.
4.2 X-ray structure determinations
Crystal data and a summary of the data collection and structure refinement are given in Table 1. The structure was solved by the PATTY option [15] of the DIRDIF program system [16]. All non-hydrogen atoms were refined [17] with anisotropic temperature factors. The hydrogen atoms were placed at calculated positions, and refined isotropically in riding mode.
Crystallographic data for 1
| Compound | 1 |
| Crystal colour | dark purple-black |
| Crystal shape | regular fragment |
| Crystal size | 0.18 × 0.17 × 0.10 mm |
| Empirical formula | C37H53AlClN3 |
| Formula weight | 602.25 |
| Temperature | 293(2) K |
| Radiation / Wavelength | MoKα (graphite monocrystal)/0.71073 A |
| Crystal system, space group | Orthorhombic, Pbca |
| Unit cell dimensions | a, α = 17.628(4) Å, 90° |
| 20087 reflections | b, β = 18.311(7) Å, 90° |
| 5.020 < θ < 24.990 ) | c, γ = 22.904(11) Å, 90° |
| Volume | 7393(5) Å3 |
| Z, Calculated density | 8, 1.082 Mg m–3 |
| Absorption coefficient | 0.154 mm–1 |
| Diffractometer / scan | Nonius Kappa CCD with area detector θ and ω scan [19] |
| F(000) | 2608 |
| Theta range for data collection | 5.02 to 24.99° |
| Index ranges | –20 ≤ h ≤20, –21 ≤ k ≤ 21, –27 ≤ l ≤ 25 |
| Reflections collected / unique | 76729/6452 [R(int) = 0.1016] |
| Reflections observed | 3930 ([I0 > 2 σ(I0)]) |
| Absorption correction | SADABS multiscan correction [18] |
| Refinement method | Full-matrix least-squares on F2 |
| Computing | SHELXL-97 [15] |
| Data / restraints / parameters | 6452 / 0 / 402 |
| Goodness-of-fit on F2 | 1.032 |
| SHELXL-97 weight parameters | 0.0543, 6.1337 |
| Final R indices [I > 2 σ(I)] | R1 = 0.0600, wR2 = 0.1269 |
| R indices (all data) | R1 = 0.1170, wR2 = 0.1529 |
| Largest diffraction peak and hole | 0.276 and –0.299 e A–3 |
The structure and atomic numbering for 1 are shown in Fig. 1. Selected bond distances are shown in Fig. 2. Geometrical calculations [18] revealed neither unusual geometric features, nor unusual short intermolecular contacts. The calculations revealed no higher symmetry and no (further) solvent accessible areas. Carbon atom C2 is disordered over two distinct positions, C2A and C2B, with occupancy factors of 0.40 and 0.60, respectively. Some other end-of-chain carbon atoms have rather large thermal displacement parameters. Because only a few of them (not even the ones with the largest displacements) could split up in the least squares refinement, it was decided not to split them up at all. Crystals of 5 were of poor quality. The (partial) X-ray structure (details given in supplementary information) was sufficient to establish its connectivity, but does not warrant publication or inclusion in the CCDC.
5 Supplementary material
Crystallographic data (excluding structure factors) for 1 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 224139. Copies of available material can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0) 1223-336033 or e-mail: teched@chemcrys.cam.ac.uk). More details can be found in the supporting information.
6 Calculations
All calculations were carried out with the Turbomole program [21,22] coupled to the PQS Baker optimiser [23,24]. Geometries were fully optimised as minima or transition states at the unrestricted bp86 [25,26]/RIDFT [27] level using the Turbomole SV(P) basisset on all atoms. All stationary points were characterized by vibrational analyses (numerical frequencies); ZPE and thermal (enthalpy and entropy) corrections (1 bar, 273 K) from these analyses are included. All energies mentioned in text and tables are free energies.