

1 Introduction
Since the first synthesis of metallocarboranes [1], cobalt dicarbollides found important applications in the field of radioactive-ion partitioning from nuclear waste solutions [2–6]. These inorganic anions are chemically stable and hydrophobic and, when added with extractant molecules (e.g., PEG or crown ethers [7,8], phosphoryl ligands [9,10], malonamides [11], calixarene derivatives [7,12,13]), enhance their extraction efficiency. Dicarbollides can be used directly or grafted onto chelating moieties like CMPO or polyethers [14–23]. There is so far no good explanation on the mechanism of dicarbollide-‘catalysed’ ion extraction.
In this paper, we report a theoretical study on the effect of CCD– anions (CCD– = chlorinated cobalt dicarbollide [Co(C2B9H8Cl3)2]–; see Fig. 1) on the extraction of M3+ lanthanide or actinide cations by a calix [4]arene-CMPO ligand (noted L; see Fig. 1) developed by V. Böhmer et al. [24]. This calixarene with four CMPO functions at the wide rim and four hydrophobic O–C10H21 chains at the narrow rim proved to extract actinides and lanthanide cations more efficiently than does ungrafted CMPO [24,25], and addition of CCD– anions enhances the extraction [26]. In order to gain microscopic insights into this synergistic effect, we decided to simulate these species at a water–‘oil’ interface [27], where ‘oil’ is modelled by chloroform, with the aim of depicting the distribution of the free ligands L, of a typical complex EuL3+, of CCD– species, and of the Eu(CCD)3 salt in different combinations. By comparing systems without CCD– and with CCD– at different concentrations we hope to understand the effect of CCD– on the extraction process. pH neutral and nitric acid solutions are considered. Most simulations consider the anionic form CCD–, but the neutral CCD0 form is also considered for comparison. Some of these solutes are simulated at a preformed interface for time scales of ~1 ns. An important issue in modelling concerns the sampling of the relevant states of the system and, in order to avoid being trapped near a starting configuration, we also performed mixing/demixing MD experiments, where ‘random’ mixtures of water, oil and the solute are first prepared, and their spontaneous evolution is simulated at 300 K. It is important to assess (i) the extent of water/oil separation, and (ii) the behaviour of the CCD– anions and of the complex and L molecules: will they finally in the organic phase where they are more soluble, or concentrate near the interface? What is the driving force for migrating from the interface towards the oil phase? What is the effect of CCD– on the lanthanide cation distribution?
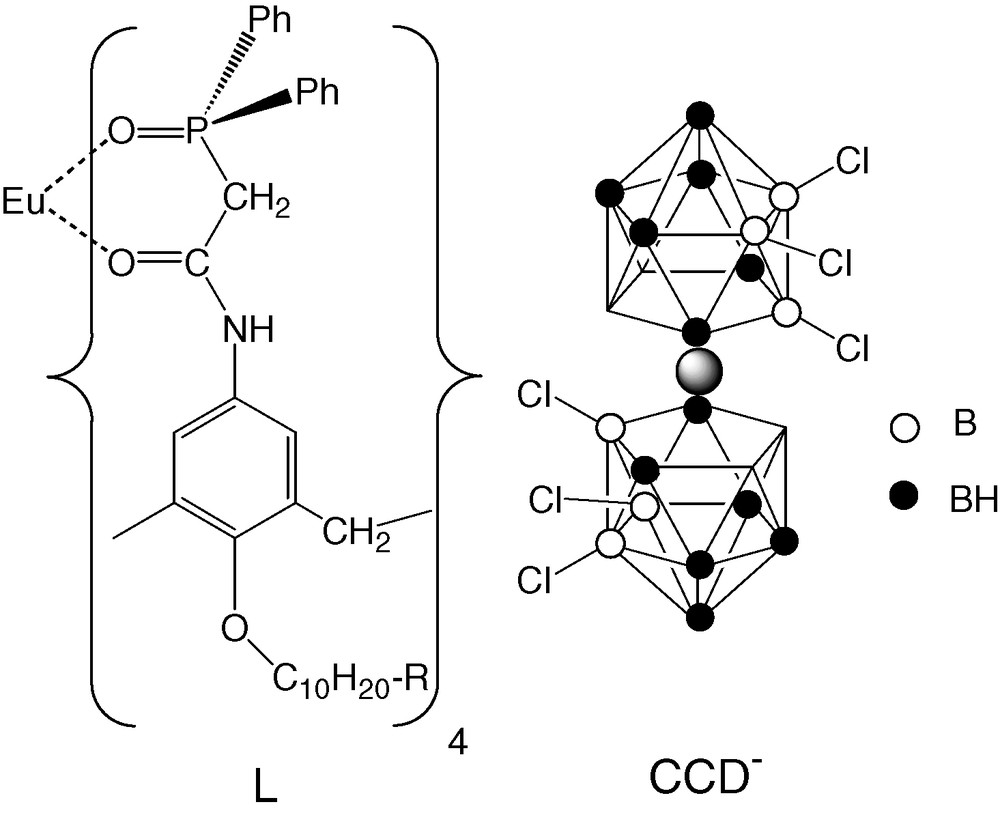
The simulated EuL3+ complex, the LCCD ligand and CCD– dicarbollide anion. R = H in L and CCD– in LCCD.
2 Methods
Molecular dynamics (MD) simulations have been performed at the water–chloroform interface using the modified AMBER5.1 software [28] with the following representation of the potential energy U:
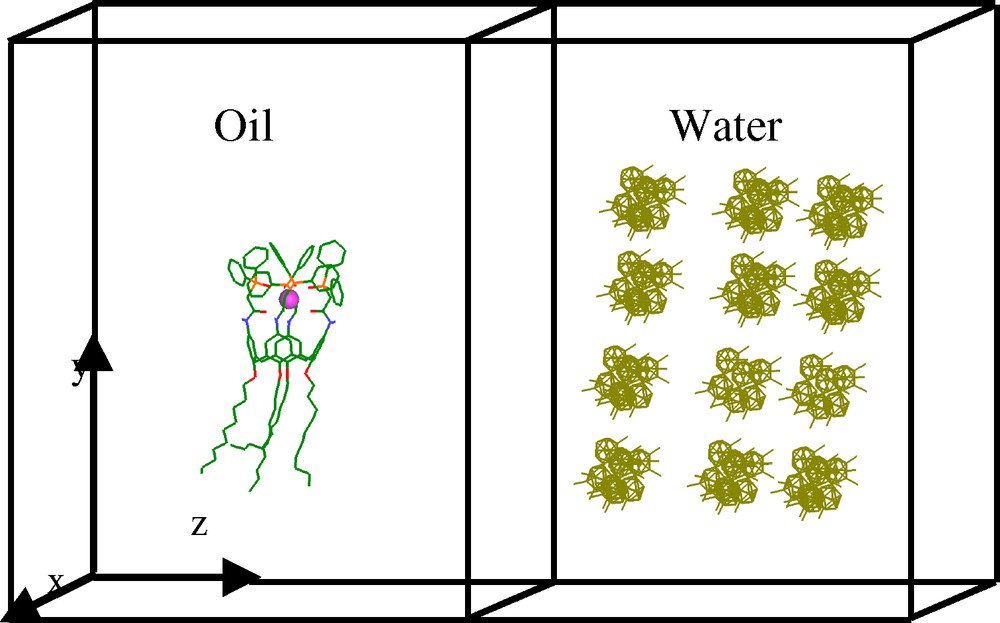
Schematic representation of the chloroform/water interface (solvent molecules not shown) with 60 CCD– and one EuL3+ complex.
Characteristics of the simulated systemsa
| Systems | Nchlor + NWAT | Box (Å3) | Time b (ns) | |
| Aint | 9 L | 874 + 3248 | 55 × 55 × (46 + 43) | 1.7 |
| Aort | 9 L | 860 + 3593 | 55 × 55 × (48 + 41) | 3.0 |
| Adem | 9 L | 860 + 3593 | 55 × 55 × (48 + 41) | 0.1/1.4/3.0 |
| B | EuL(NO3)3 | 1002 + 4286 | 55 × 55 × (46 + 43) | 1.0 |
| C | EuL3+, 4 CCD–, 1 H3O+ | 999 + 4275 | 55 × 55 × (46 + 43) | 2.0 |
| D | EuLCCD–, 1 H3O+ | 988 + 4248 | 55 × 55 × (46 + 43) | 1.0 |
| E | 13 Eu3+, 40 CCD–, H3O+ | 917 + 4052 | 55 × 55 × (46 + 43) | 0.1/1.5/1.5 |
| F | EuL3+, 24 CCD–, H3O+, 50 ‘acid’ a | 921 + 3855 | 55 × 55 × (46 + 43) | 0.1/1.3/1.8 |
| G | EuL3+, 44 CCD–, 41 H3O+, 50 ‘acid’ a | 828 + 3625 | 55 × 55 × (46 + 43) | 0.1/1.0/1.5 |
| H | EuL3+, 4 CCD–, 40 CCD0, 1 H3O+, 50 ‘acid’ a | 873 + 3670 | 55 × 55 × (46 + 43) | 0.1/1.5/1.6 |
a acid = HNO3, H3O+, NO3–.
b The times are given for equilibration/mixing/demixing, respectively.
The mixing/demixing MD simulations started from a system equilibrated at the water–‘oil’ interface. The mixing was achieved by increasing the temperature to 500 K and scaling down the electrostatic interactions by a factor of 100. Constraints of 10 kcal mol–1 were imposed on the Eu–O distances of the EuL3+ complex in order to prevent its dissociation during the heating process and the first 50 ps of demixing, which were simulated by resetting the temperature to 300 K and the dielectric constant to 1.0. The coordinates were saved every 0.25 ps, and analysed using the MDS and DRAW software [41]. The position of the interface was defined as the interaction between the water and oil density curves [42].
3 Results
In the following, we mainly focus on the distribution of the solutes near the interface, with the main aim to understand under which circumstances the lanthanide complex looses contact with the water phase and can be considered, at the microscopic level, as extracted to the ‘oil’ phase. As concerns the L ligand and its EuL3+ complex, they are quite flexible, but their cone moiety retains an approximate fourfold axis symmetry, with respect to which ‘axial’ and ‘equatorial’ positions can be defined. Unless otherwise specified, all results are obtained with the 15 Å + RF treatment of electrostatics.
3.1 The free L calixarene ligand, the EuL3+ complex and the CCD– anions are surface active
According to the MD simulations, the free ligands, the EuL3+ complex and the CCD– anions adsorb at the water/oil interface. This can be seen first in system A, when nine L ligands are simulated at a preformed interface starting with a grid of 3 × 3 L parallel (A//) or perpendicular (A⊥) at the interface (Fig. 3), as well as from a demixing simulation (Ademix; see Fig. 4). The final ligand distribution somewhat depends on the starting state, and can be a single monolayer at the starting interface (A//), or dilute (4 +5 ligands) onto the two equivalent interfaces (Ademix), or form a mixture of interfacial + oil solution (A⊥). These differences relate to the 3D-boundary conditions imposed to the system and to the truncation of ‘long-range interactions’, which implies that there is no driving force for the molecules beyond 15 Å from the interface to be attracted by the latter. In most cases, the L molecules have their ‘symmetry axis’ perpendicular to the interface, with CMPO moieties on the water-side and the O-alkyl chains on the oil-side of the interface. An important driving force seems to be the hydration of the phosphoryl oxygens that are hydrogen bonded to water molecules. According to an energy components analysis on the Ademix system, each ligand is much more attracted by water than by chloroform (ΔE = –240 and –120 ± 5 kcal mol–1, respectively).
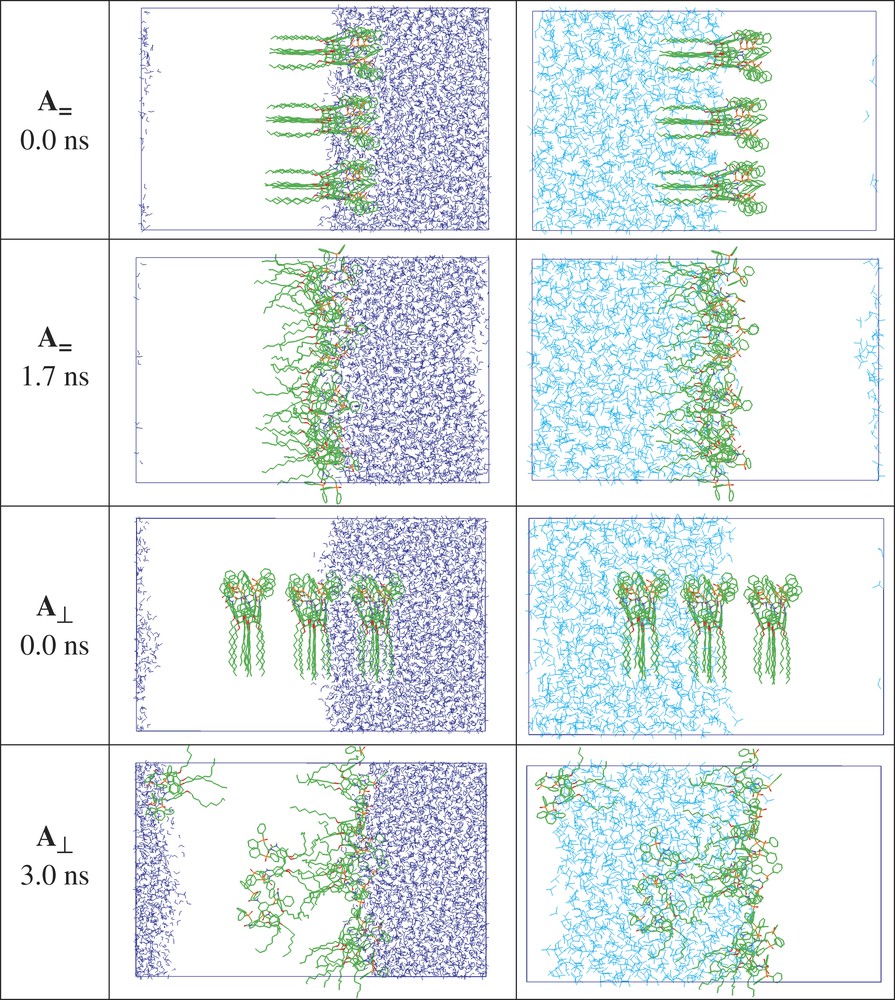
System A (9 L) at the interface, starting with a 3 × 3 grid parallel or perpendicular to the interface. Snapshots at the beginning (0 ns) and at the end of the simulation. The water and chloroform solvents are shown side by side for clarity.

System A (9 L calixarenes) at the beginning (0 ns) and the end (3 ns) of MD demixing simulation. The water and chloroform solvents are shown side by side, instead of superposed, for clarity.
The EuL3+ complex similarly adsorbs at the interface, be it neutralized by 3 NO3– or 3 CCD– anions (systems B and C; Fig. 5). Its cationic site has no direct contact with the counterions, but interacts with the interfacial water molecules, with different patterns, though. In the EuL(NO3)3 system B, the cation is fully encapsulated within the four CMPO arms and shielded from the medium. One thus finds ‘second-shell’ water molecules in equatorial position and one nitrate anion around EuL3+. This contrasts with the EuL(CCD)3 system C, where the cation is ‘axially coordinated’ to water molecules that are in endo and exo positions. One observes different behaviours for the counterions. Nitrates are hydrophilic and two of them sit finally in water, while the CCD– dicarbollides spread onto the interface, where they are more attracted by water than by oil (–50 and –15 ± 4 kcal mol–1, respectively). A similar situation is observed when the four CCD– anions are covalently linked to the complex via the O-(CH2)10 connectors (system D; Fig. 5). These chains are long enough to allow for the CCD– groups to adsorb at the interface as in the absence of linker, while the complexed cation is also hydrated by second-shell ‘equatorial’ water molecules.

Simulation of systems B (EuL(NO3)3), C (EuL3+, 4 CCD–, 1 H3O+) and D (EuLCCD–, 1 H3O+). Snapshots at the beginning (0 ns) and at the end of the simulation. The water and chloroform solvents are shown side by side for clarity. A zoom of the complex is shown on the right-hand side.
The distribution of the Eu(CCD)3 salt was studied (system E with 13 Eu3+, 40 CCD–, H3O+ ions; see Fig. 6) from a mixing-demixing MD experiment. At the beginning of the demixing stage (0 ns), all solvent molecules and ions were randomly dispersed. Rapidly, however, water and oil separated, forming two phases that finally (at 2.5 ns) delineated two interfaces. Not surprisingly, nearly all CCD– anions adsorbed on the water surface, as in simulation C. The most interesting result concerns the distribution of the Eu3+ cations (Fig. 6). Generally, such hard and highly hydrophilic cations are ‘repelled’ by aqueous interfaces, preventing therefore their capture by interfacial ligands. This has been observed computationally for, e.g. KCl [43], UO2(NO3)2 [44] or EuCl3 salts, for which the average cation density is nearly zero at the interface, but peaks in the bulk aqueous phase, thus following pictures inferred from surface tension measurements. In the E system, the adsorbed CCD– anions create a negative potential that attracts the cations as hydrated Eu(H2O)93+ species, some of which are close to CCD– anions at the interface. In order to check whether such an unexpected distribution is not an artefact resulting from the treatment of long-range forces, we decided to rerun the E demixing simulation with the PME Ewald summation method (EPME), instead of the 15 Å + RF treatment. The final result turned out to be nearly identical (Fig. 6), confirming the peak of lanthanide cation concentration near the interface, induced by the CCD– counterions.
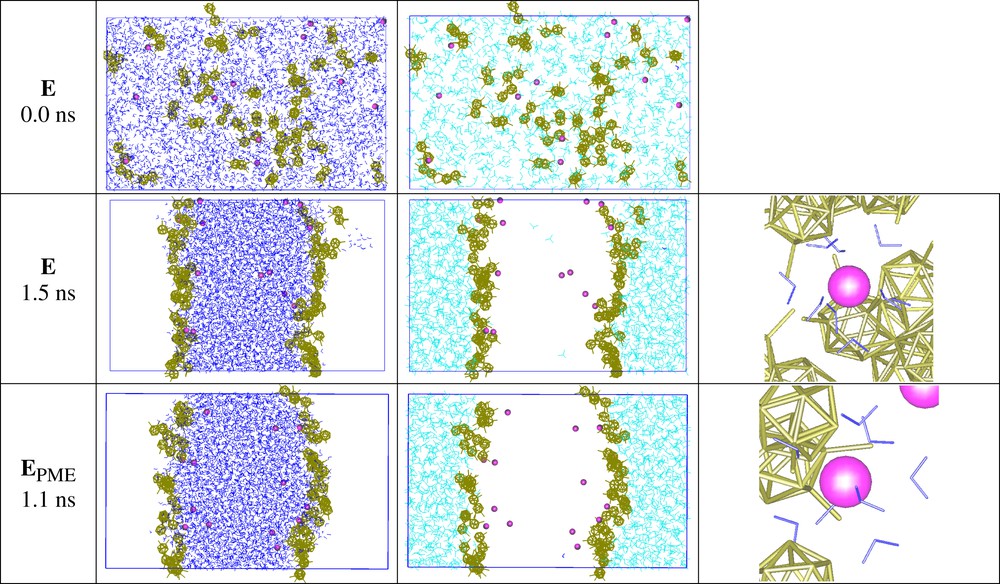
Demixing simulations of system E. Initial state (0 ns; top) with ‘randomly mixed’ solvent molecules and ions, and final state after 2.5 ns of demixing with the 15 Å + RF method (middle), or with the Ewald PME method (bottom). The water and chloroform solvents are shown side by side, instead of superposed, for clarity. Zooms of interfacial Eu(H2O)93+ complexes are shown on the right-hand side.
The fact that all key partners (the free ligand L, its lanthanide complex and the uncomplexed cation) concentrate in the interfacial region strongly suggests that the cation capture and recognition processes take place at the interface. A subsequent question, in the context of ion extraction, is to understand how the complexes desorb from the interface and migrate to the oil phase. Surface activity implies that these species concentrate at the interface, and this feature has to be taken into account in the simulations. This is addressed in the next section. Since the microscopic concentrations are unknown from experiment, we will consider an excess of dicarbollides with respect to the complex. We also take into account the effect of acidity, via a comparison of acidic vs. ‘pH neutral’ systems.
3.2 Demixing simulations on the water/oil/CCD–/EuL3+ acidic mixtures
The effect of the CCD– anion concentration can be seen in demixing simulations of water/oil mixtures containing one EuL3+ complex, a large amount of nitric acid (about 4 molar, modelled by 50 HNO3, NO3–, H3O+ species) and of CCD– anions (systems F with 24 CCD– and G with 44 CCD– anions). It can be seen in Fig. 7 that, in both systems, water, oil and the solutes are ‘randomly mixed’ at the beginning of the simulation (0 ns). In the two cases, one observes at the end of the dynamics a nearly complete phase separation, and the formation of two interfaces. Phase separation is somewhat slower in G than in F (2.3 ns and 1.5 ns, respectively), in keeping with the larger CCD– concentration in G. There is finally no chloroform in the water phase, and no water in ‘oil’, the only exception being a small water droplet near the interface in F. In both systems, most of the CCD– anions adsorb at the two interfaces, and the EuL3+ complex adsorbs at one of them. The acid components (HNO3, NO3–, H3O+) sit in the bulk aqueous phase, as expected. In contrast to observations made with pure nitric acid only [45], one does not find HNO3 species at the interface, while the H3O+ ions prefer to be solvated by bulk water than to concentrate near the interfacial CCD– anions. In both systems, the interface onto which EuL3+ adsorbs is much less planar and regular than the other one. As far as ion extraction is concerned, there is an important effect of CCD– concentration, though. In system F, the interfaces are not fully covered by the anions, and the interfacial EuL3+ complex remains in contact with water. The dilution of CCD– anions at the interface somewhat prevents the local neutralization of the complex which sits rather ‘tangential’ to the interface. At higher CCD– concentration (G), the microenvironment of the complex differs, due to the local saturation of the interface by the CCD– anions. The EuL3+ complex thus moved somewhat to the oil phase, followed by a few dragged water molecules and can be thus considered to sit on the extraction pathway.
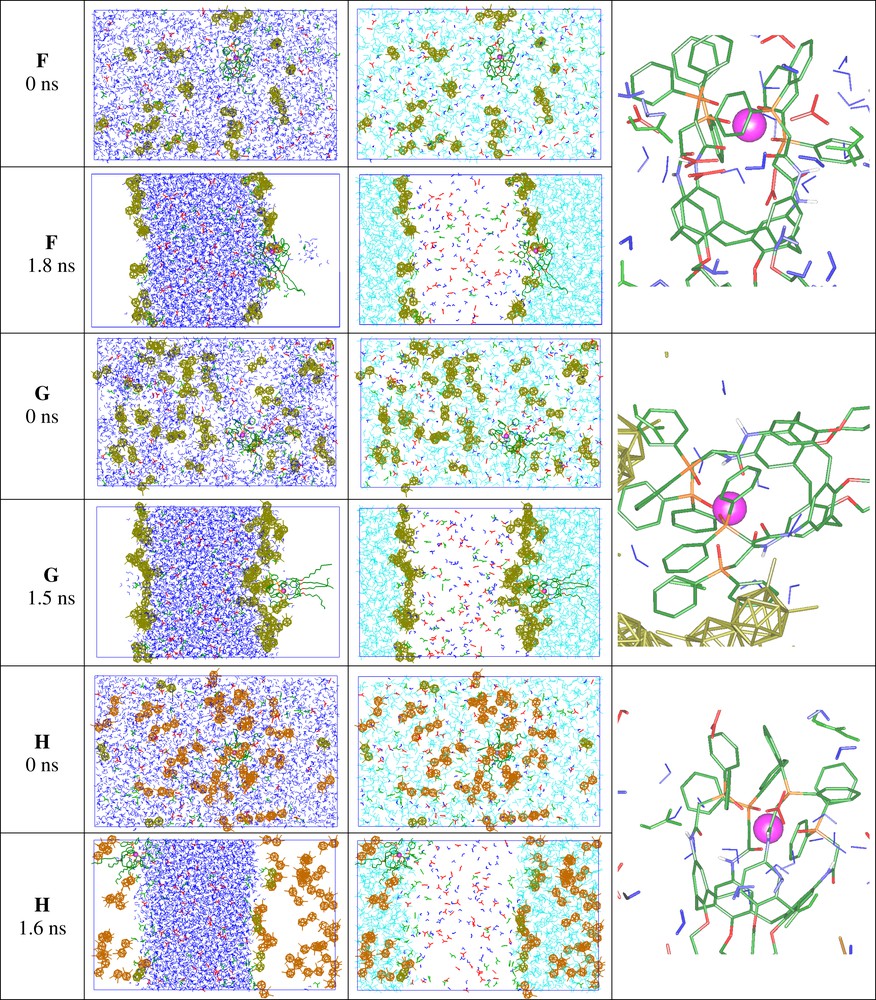
Demixing simulations of systems F, G, H. Snapshots at the beginning (0 ns) and at the end of the simulation with water and chloroform shown side by side, instead of superposed, for clarity. A zoom of the complex is shown on the right-hand side.
The importance of electrostatics on the distribution of CCD– anions can be seen from another simulation on a system analogous to G, in which the dicarbollides were ‘neutralized’ (system H; see Fig. 7). This was achieved by replacing 40 CCD–, H3O+ by 40 neutral CCD0 species, which mimic the neutral form of the CCDH acid. A mixing–demixing simulation of H also led to the separation of water and oil phases, as observed with system G, while nitric acid also solubilized in water. The distribution of ‘dicarbollides’ dramatically differs, however, as most of the CCD0 molecules are now immersed in oil, instead of adsorbing at the interface, as a result of their hydrophobic character and their zeroed electrostatic interactions with water. As a consequence, the EuL3+ complex is less shielded from the interface than it was with CCD– anions, and is strongly attracted by water (–360 kcal mol–1). It can thus be hardly extracted to oil. Insights into the effect of acid were obtained by performing some simulations without acid [46]. The final situation is quite similar, as far as the interfacial activity of CCD– anions and their role on the extractability of the EuL3+ complex are concerned. As nitric acid sits in the water phase, it is not surprising to find that the distribution of neutral CCD0 is also quasi acid-independent.
4 Conclusion
We report molecular dynamics simulations on several interfacial systems involved in the lanthanide ion extraction by a functionalised calix[4]arene, with the aim to understand why dicarbollide anions facilitate (‘catalyse’) the process. These are part of a series of ‘MD experiments’ that we performed on electrolytes, ligands, complexes in different combinations at liquid–liquid interfaces, in relation with the ion extraction process. Like any theoretical approach, the models (e.g., size of the solvent box, force field) and simulation methods result from a compromise between feasibility (in terms of computer time and human time) and ‘accuracy’. In previous papers, we addressed important methodological issues like the water model (e.g., TIP3P versus TIP5P versus polarisable models), the choice of thermodynamic conditions (N,P,T versus N,V,T ensemble and control of temperature), the treatment of electrostatics (polarization effects, truncation of the interactions at the boundaries, ‘long-range’ interactions), the sampling and equilibration of the system [47–51]. The results presented here are consistent with these studies and allow us to understand the synergistic role of CCD– anions at the interface.
A first important result is the high surface activity of the CCD– anions, which contrasts with the inactivity of their corresponding neutral fictitious CCD0 analogues. The free ligand L and its EuL3+ complex are also surface active, which strongly suggests that the cation capture by L takes place at the interface, and this is can be again promoted by CCD– anions. Generally, hard ions are instead ‘repelled’ by the interface, but the interfacial film formed by CCD– anions creates a negative potential, which attracts Eu3+ cations and should therefore ‘catalyse’ their capture by the ligands.
The interfacial activity of the calix[4]arene L and its complexes is consistent with expectations based on their amphiphilic character and analogies with other extractant molecules [52]. As far as CCD– anions are concerned, this can be more surprising, as these species are quite ‘spherical’ and lack amphiphilic topology. Surface activity has been attributed to the fact that these hydrophobic ions still enjoy important attractive interactions with water at the interface, while avoiding paying for the cavitation energy in water [53]. Their interfacial activity is consistent with previous results obtained with tetrahedral hydrophobic ions (AsPh4+ and AsBPh4–) as well as with spherical hydrophobic S+ and S– ions of similar sizes [43,48,51]. It is also consistent with experimental results obtained at the water–air interface, which bears analogies with the water-oil interfaces [54], and with surface tension measurements according to which the surface tension decreases with the larger ions [55–57]. The interfacial activity of CCD– anions is also of interest in the context of the Hofmeister series (SO42– > F– > Cl– > I– = ClO4– > SCN–) that was empirically established from denaturation studies of proteins by salts [58]. As concerns simulation results, it has been recently noted that, in the halide series, adsorption at aqueous surfaces increases with the anion size and polarisability, i.e. from F– to I– [59–61]. The CCD– anions being still bigger than I– should sit at the right-hand side of the Hofmeister series and should be therefore most surface active. A still increased surface activity can thus be anticipated if polarization of the solvents and the anions are explicitly accounted for [59,62–64].
Another important issue concerns the driving force for diffusion of the complexes from the interface to oil, and the anion concentration again plays a major role. At high concentrations, CCD– saturate the interface, attract the EuL3+ complex, which becomes locally neutralized, a requisite for extraction. The complex thus interacts less with water at the interface than it does in the bulk solution. Extraction can be also induced or facilitated by other factors, like salting out effect, and reduction of the interfacial area. We note that our simulations deal with micro-interfaces (of a few nanometres wide), which can represent, for instance, parts of macro-interfaces or of the surface of water-in-oil droplets. Upon collapse of droplets, the interfacial area decreases and the ligands, complexes and synergistic anions should be expelled from the interface to the phase where they are most soluble, i.e. to oil.
Another issue concerns the microscopic nature of the extracted complex. We modelled complexes of 1:1 stoichiometry, for simplicity. The reality may be more complicated, however, due to the possible formation of supramolecular assemblies ranging from aggregates, oligomers, or more complex organized systems as suggested by NMR [65], diffraction measurements [66,67], and X-ray structures of analogues [68]. Our simulations, however, strongly point to the importance of interfacial phenomena and to the interfacial role of anions like CCD–. Beyond liquid–liquid extraction, these data are also relevant for other aqueous interfaces, involved, for instance, in electrochemical processes [69] or transfer through lipidic membranes [70]. They also bear some analogies with the surface of charged micelles, for which important counterion effects have been noticed. For instance, multivalent counterions, such as Al3+ and Ca2+ are known to be much more effective promoters of micelle growth than monovalent counterions such as Na+ [71], and this is consistent with the neutralization of the CCD– layer by Eu3+ cations observed in our simulations of the Eu(CCD)3 salts. Generally speaking, this study illustrates the increasing role of molecular dynamics simulations to depict at the molecular level the time evolution and statistical distribution of heterogeneous and complex systems, of growing importance in chemistry, biology, and physics.
Acknowledgements
The authors are grateful to EEC (contract F1KW-CT2000-0088) and PRACTIS for support, and to IDRIS, CINES and ‘Université Louis-Pasteur’ for allocation of computer resources.