

1 Introduction
The design of synthetic pathways to attain functional materials continues to be a challenge for inorganic chemists. In this context, a great effort has been dedicated to the development of rational synthetic routes to obtain novel polynuclear compounds of tunable dimensionality. Engineering molecular-based magnetic materials [1–5] requires building molecular networks in the crystalline phase. This implies identifying appropriate tectons, that is, active molecular building units the constitution and assembling properties of which favor the expected network dimensionality [6–7]. Along this line, many recent reports have focused on the synthesis and structural characterization of polymeric transition metal, rare earth and hetero metal compounds using dicarboxylates of different length as ionic molecular tectons (see for example [8–14]). Depending on the length of the dicarboxylate chain –[O2C(CH2)nCO2]– a large variation in coordination modes, which can coexist, can be constructed. Binding ways include chelating, bidentate bridging or chelating bridging modes. They help towards the formation of clusters, polymeric structures and open-frameworks which give rise to materials with cooperative magnetic interactions of different dimensionality. Many authors have claimed [13–19] that hydrothermal synthesis is necessary to produce open-framework structures. However, a gel diffusive technique has also been successfully employed to obtain some rare earth dicarboxylates with polymeric structures [11–12,20]. Some authors have prepared transition metal carboxylates under ambient conditions [21–23]. We report here the synthesis and crystal structure of [Nd2(O2C(CH2)4CO2)3(H2O)4]·6 H2O (1) obtained from solution under ambient conditions. To our best knowledge no rare earth porous adipates has been previously obtained in this way. The magnetic response of a polycrystalline sample of 1 was studied down to 14 K from AC susceptibility. To better understand the relative influence of the different factors that may cause the magnetic transition observed at 280(2) K, two other Nd adipate hydrates were produced. One was prepared varying the crystallization conditions, [Nd2(O2C(CH2)4CO2)3(H2O)2]·H2O (2), and the other by dehydration of 1, [Nd2(O2C(CH2)4CO2)3(H2O)2] (3). Possible relationship between water content and magnetic behavior is discussed.
2 Results and discussion
2.1 Crystal structures
Crystal data of 1 and 2 at 120(1) and 293(2) K are presented in Table 1. No indication of structural phase transitions within these temperatures has been observed. The structure analysis shows that compound 1 is isostructural with the La derivative obtained by Kiritsis et al. [11] and with the Ce derivative obtained by Sun et al. [12], both synthesized by gel diffusion (present unit cell is related with La and Ce derivatives unit cells by the transformation: 100/1–10/001). Fig. 1a shows the Nd(III) ions coordination and some relevant geometric features are given in Table 2. It can be seen that, in compound 1, the Nd(III) coordination sphere, comprises nine oxygen atoms, seven belonging to five adipate anions and two to water molecules. Two of the adipate ligands bind the Nd ions through a bidentate interaction, while the other three show a monodentate interaction. The adipate dianions conformations and binding modes to lanthanides have been described by several authors [16,18]. Fig. 2 shows the three different types of ligand conformations and binding modes observed in 1 and 2. As shown in Fig. 3a, along the a axis the Nd(III) ions are alternately connected by two chelating bridging carboxylates (where chelating plane, BP1′ – Table 2 displays planes labeling – departs 8.5(2)[8.51(9)]° from BP1 plane) and by two bidentate bridging carboxylates (these form an eight member ring, where the BP2′ plane departs 1.85(7)[2.1(1)]° from BP2 plane) building an infinite polymeric chain. Adjacent bridging planes (BP1 and BP2) form an angle of 36.5(2)° [35.9(1)°]. The Nd···Nd2 and Nd···Nd3 distances are 4.4767(3) Å [4.4919(6) Å] and 4.8167(3) Å [4.8546(9) Å], respectively (2: 2 – x, 1 – y, –z; 3: 1 – x, 1 – y, –z). The values corresponding to 293 K data are in brackets and evidence significant variations when going from 293- to 120-K structure. Variations in Nd–O(21)2 and Nd–O(12) distances with the change of temperature are also observable in Table 2. The infinite chains are interlinked by the anions lying approximately along the crystallographic b and c axes. They help the development of an open-framework with two set of channels, both running parallel to the Nd chains as shown in Fig. 4a. The estimated cross section of one of the sets, where the hydration and coordination waters are located, is 63 Å2. The other set of channels present a smaller cross section, 33 Å2. The adipates lying along c are on an inversion center and are linked to four Nd atoms. Following Kim and Jung [18] they can be classify as L1 (anti/anti/anti), while the adipates lying along b present the form L2 (gauche/anti/anti) and are linked to three Nd(III) ions.
Crystal data and structure determination summary for [Nd2(O2C(CH2)4CO2)3(H2O)4]·6 H2O (1) and [Nd2(O2C(CH2)4CO2)3(H2O)2]·H2O (2) at 293 and 120 K
| 1 | 2 | 1 | 2 | |
| Temperature (K) | 120(2) | 120(2) | 293(2) | 293(2) |
| Chemical formula | C9H22NdO11 | C9H15NdO7.50 | C9H22NdO11 | C9H15NdO7.50 |
| Formula weight | 450.51 | 387.45 | 450.51 | 387.45 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P | C2/c | P | C2/c |
| a (Å) | 9.1820(1) | 17.8706(4) | 9.241(1) | 17.9796(4) |
| b (Å) | 9.6870(1) | 7.8010(2) | 9.751(2) | 7.8105(2) |
| c (Å) | 10.6390(2) | 18.8326(4) | 10.661(2) | 18.8844(4) |
| α (°) | 106.807(1) | 90.000 | 106.93(3) | 90.000 |
| β (°) | 95.625(1) | 111.990(1) | 95.32(2) | 111.810(1) |
| γ (°) | 118.060(1) | 90.000 | 118.32(2) | 90.000 |
| Volume (Å3) | 768.10(2) | 2434.4(1) | 778.0(2) | 2462.1(1) |
| Dcalc (g cm–3) | 1.948 | 2.114 | 1.923 | 2.090 |
| Z | 2 | 8 | 2 | 8 |
| μ (mm–1) | 3.430 | 4.291 | 3.387 | 4.242 |
| Reflections collected with I > 2 σ(I) | 5105 | 9356 | 5177 | 9589 |
| Independent reflections | 2712 | 2753 | 4505 | 2823 |
| Parameters | 190 | 160 | 198 | 160 |
| Goodness-of-fit on F2 | 1.419 | 1.069 | 1.059 | 1.051 |
| R | 0.0226 | 0.0316 | 0.0207 | 0.0366 |
| Rw | 0.0920 | 0.0661 | 0.0462 | 0.0721 |
| Largest difference in peak and hole (e Å–3) | 0.720/–1.747 | 0.747/–1.265 | 0.939/–0.642 | 0.699/–0.921 |
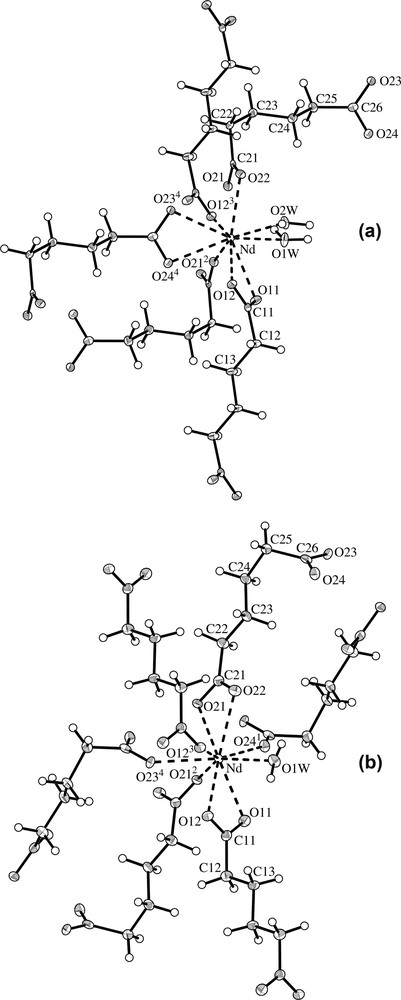
Coordination of Nd(III) ions including the labeling scheme. (a) [Nd2(O2C(CH2)4CO2)3(H2O)4]·6H2O, (b) [Nd2(O2C(CH2)4CO2)3(H2O)2]·H2O.
Selected bond distances (Å), angles (°) and planes for 1 and 2
| Atoms | Compound 1a | Compound 2a |
| Selected distances | ||
| Nd–O2W or O(24)1 | 2.463(3) [2.468(2)] | 2.399(2) [2.400(2)] |
| Nd–O(21)2 | 2.428(3) [2.418(2)] | 2.462(2) [2.462(2)] |
| Nd–O(1W) | 2.483(3) [2.481(2)] | 2.484(2) [2.486(2)] |
| Nd–O(11) | 2.536(3) [2.526(2)] | 2.507(2) [2.507(2)] |
| Nd–O(12)3 | 2.482(3) [2.475(2)] | 2.508(2) [2.513(2)] |
| Nd–O(23)4 | 2.573(3) [2.574(2)] | 2.508(2) [2.519(3)] |
| Nd–O(24)4 | 2.542(3) [2.543(2)] | |
| Nd–O(12) | 2.732(3) [2.757(2)] | 2.523(2) [2.533(2)] |
| Nd–O(21) | 2.528(2) [2.538(2)] | |
| Nd–O(22) | 2.461(3) [2.454(2)] | 2.581(2) [2.586(2)] |
| Selected angles | ||
| Nd–O(12)–Nd3 | 118.3(1) [118.21(6)] | 114.09(8) [113.91(9)] |
| Nd3–O(12)–C(11) | 149.4(3) [150.2(2)] | 147.0(2) [146.7(2)] |
| O(12)–C(11)–O(11) | 119.1(4) [119.3(2)] | 118.1(3) [118.9(3)] |
| C(11)–O(11)–Nd | 100.6(2) [101.6(1)] | 94.5(2) [94.8(2)] |
| Nd–O(21)2–Nd2 | 118.54(5) [118.21(6)] | 108.0(1) [107.54(1)] |
| O(21)–C(21)–O(22) | 121.9(4) [122.0(2)] | 118.9(3) [119.0(3)] |
| C(21)–O(21)–Nd2 | 160.6(3) [161.7(2)] | 144.4(2) [145.0(2)] |
| C(21)–O(22)–Nd | 113.3(3) [115.2(1)] | 93.6(2) [93.7(2)] |
| C(26)1–O(24)1–Nd2 | 142.7(2) [142.8(2)] | |
| Nd–O(23)1–C(26)1 | 132.5(2) [132.3(2)] | |
| O(23)–C(26)–O(24) | 120.1(4) [120.2(2)] | 125.2(3) [125.4(3)] |
| Selected planes notation | ||
| BP1: Nd, O(12), Nd3, O(12)2; BP1′: Nd, O(11), C(11), O(12) | ||
| BP2: Nd, O(21), Nd2, O(21)2 ; BP2′: Nd, O(22), C(21), O(21) | ||
| BP3: Nd, O(23)4, C(26)4, O(24)4, Nd2 |

Observed adipate ligand conformations. In L1 the two ends show a chelating bridging arrangement, in L2 the top end shows a chelating arrangement and the bottom end a bidentate bridging one, and in L2′ the top end shows a bidentate bridging arrangement and the bottom end a chelating bridging one.
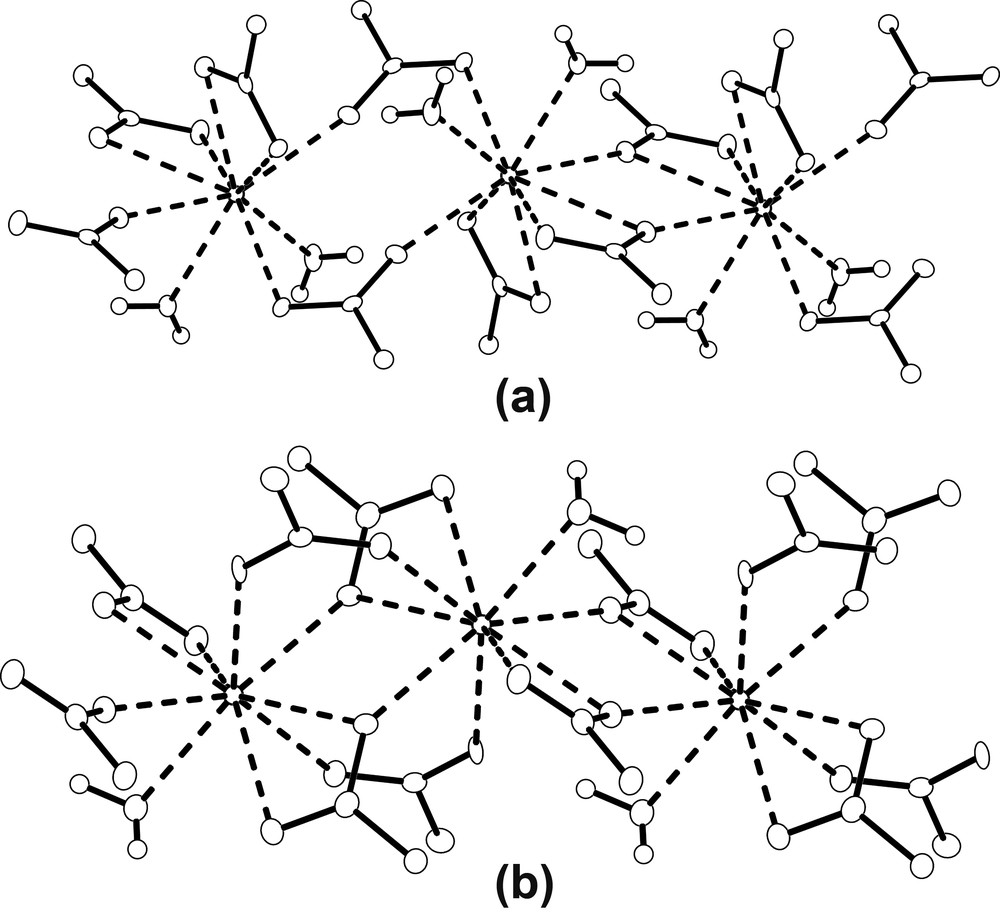
Schematic drawing of a fragment of the chains showing the bridging groups of (a) compound 1, (b) compound 2.
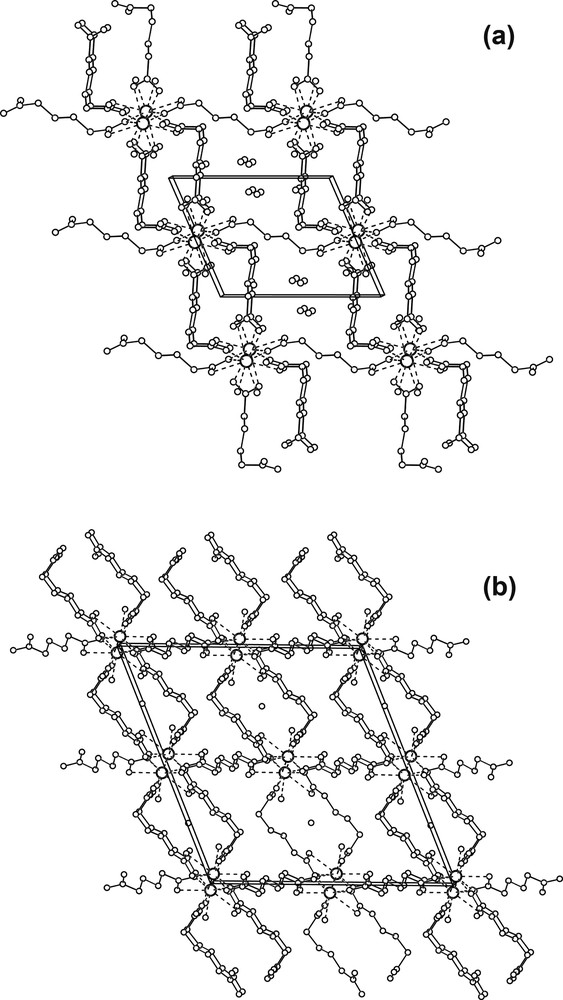
Projection of the chains packing: (a) compound 1 along a (c horizontal and b vertical), (b) compound 2 along b (c horizontal and a vertical).
In compound 2 Nd(III) ions are also coordinated by nine oxygen atoms, eight belonging to six adipate anions and one to a water molecule, as shown in Fig. 1b. Some selected bond distances and angles are given in Table 2. The coordination sphere of the Nd(III) cations consists of one oxygen atom from a water molecule, four oxygen atoms belonging to two adipate ligands, which link the Nd ions through a bidentate interaction, and four oxygen atoms belonging to four adipate ligands connected to the Nd ions through a monodentate interaction. Like in compound 1, the Nd(III) cations are linked to form infinite chains along the b axis. As shown in Fig. 3b, adjacent cations in the chain are alternately linked in two different ways: one in which two carboxylate groups in a chelating bridging mode (the chelating plane, BP1′, is out of the bridging plane BP1 24.0(1)[24.2(1)]°) are involved, and the other comprising four carboxylates, two bridging chelating (the chelating plane, BP2′, is out of the bridging plane BP2 21.3(3)[21.0(1) 1)]°) and two bidentate bridging. The last two bridging groups are lying in a plane (BP3) which is almost perpendicular to BP2, 89.96(7)° [88.4(2)°]. The angle between BP1 and BP2 in 2 is 38.8(1)°. Four carboxylate groups bridging two Nd(III) ions in a similar way have also been observed by Legendziewcz et al. in [Nd(Glut)(H2O)4]Cl·2 H2O [11]. Nd···Nd2 and Nd···Nd3 distances are 4.0360(3) Å [4.0335(3) Å] and 4.2211(2) Å [4.2295(2) Å], respectively (2: 2 – x, 2 – y, 1 – z; 3: 2 – x, 1 – y, 1 – z). These distances are shorter than those observed in 1. Unlike in 1, the Nd…Nd distances, as well as the Nd–O(21)2 and Nd–O(12) distances and the Nd–O(21)2–Nd2 and Nd–O(12)–Nd3 angles, do not change significantly with temperature (see Table 2). As illustrated in Fig. 4b, the Nd chains, like in compound 1, are interlinked by anions lying along the a and c crystallographic axes, building a porous structure with channels running parallel to the chains, where the hydration and coordination waters are located. The estimated channel cross section in this compound is 34 Å2, similar to that of the smaller channel in 1. The anions lying along a present the form L1, and are coordinated to four Nd atoms, while those lying on c adopt the L2′ form (gauche/anti/anti) and are coordinated to four Nd(III) cations.
X-ray powder diffraction analysis and DTA and TG studies of 1 [24,25] have shown (in accordance with Kiritsis et al. [11] study of a La derivative) that this compound can be dehydrated, in a reversible way, to get compound 3. The comparison of the porous structures found in 1 and 2 shows that compound 2 does not display two different sets of channels, as found in compound 1. Probably the higher synthesis temperature used to obtain 2 does not favor the retention of hydration water and helps the interactions between adipates, thus preventing the formation of large channels. Then, it seems reasonable to propose that, depending on the environmental conditions; the adipate anions may modify their conformation and thus influence the size of the channels. In 1 the extended L1 conformation favors the formation of large channels, which can then accommodated all water molecules, while in 2 the more contracted L2′ conformation induces a restriction on the cross section of one of the channels thus hindering the water access. Even though we could not obtain single crystals of 3 suitable for structure analysis, we can infer that in this dehydrated compound both sets of channels are narrowed. This model is in agreement with the specific area reduction found by Kiritsis et al. [11] when going from [La2(O2C(CH2)4CO2)3(H2O)4]·6H2O (isostructural to 1) to La2(H2O)2(C6H8O4)3 (analogue of present compound 3).
2.2 Magnetic susceptibility study
The magnetic behavior of complexes 1–3 are shown in Fig. 5 where the real part of the AC magnetic susceptibility (χ′), its inverse (χ′–1) and the susceptibility temperature product (χ′T) are plotted versus the temperature. The analysis of χ′–1 shows an overall departure from the Curie–Weiss law in the three compounds. The change of χ′–1 slope observed at TC = 280(2) K, along with the maximum found at that temperature in χ′T indicates a magnetic transition. The existence of a transition is consistent with the DSC data [24,25]. A linear fit of χ′–1 above TC, assuming a paramagnetic-like behavior, yields Weiss temperatures of 254(2), 155(2) and 130(2) K for compounds 1–3, respectively, thus indicating the existence of short order ferromagnetic exchange interactions. Below TC, both χ′–1 and χ′T decrease with the temperature and χ′–1 curves deviate from linearity for the three compounds. The dependence of 1 with temperature is dissimilar to that observed in compounds 2 and 3. The absence of a maximum in χ′ up to the lowest temperature reached does not allow to establish the existence of long-range antiferromagnetic interactions. The observed deviation of χ′–1 from linearity may be due to the unidimensionality of the superexchange paths connecting the Nd(III) cations, observed in 1 and 2 and described above. However, splitting of the free-ion ground state under the influence of the crystal field (CF) would also explain the present results [1]. Different rare earth systems – where no interaction between rare earth ions are expected – show deviations in χ′–1 at low temperature similar to the present ones. Departure form linearity has been explained by Cascales et al. [26,27] as due to CF effects. Theoretical calculations on model low dimensional molecular-based magnetic materials performed by Kaneyoshi and Nakamura [28] predict a departure of χ′–1 from linearity at low temperature, but only for some values of the CF parameters. From the results of the mentioned authors it seems that CF effects might be the main cause of the departure from the linearity at low temperature. However, evidences are contradictory, e.g. splitting of the ground state of free Nd(III) ions by CF allowed Legendziewcz et al. to explain the optical behavior of [Nd(Glut)(H2O)4]Cl·2 H2O [11], where Nd(III) ions present a nine coordination and super exchange paths between them comparable to those found in 1 and 2. In spite of that no χ′–1 departure from linearity has been observed in that compound. Therefore, it is clear that theoretical calculations and more experimental results are necessary to fully understand the magnetic response of 1–3. Some experiments are already underway. It is worthwhile to mention that AC magnetic susceptibility data obtained from partially rehydrated samples of 3 have shown a correlation between the increase of water content and the amplification of the susceptibility value [24]. This can be assigned to different causes, such as changes in CF parameters, modifications of inter-chain interactions and hydrogen bonding, as found by Price et al. [29] in some Mn(III) complexes, or contribution of vibronic interactions induced by waters as those observed by Dalosto et al. [30] in copper-doped bis(d,l-histidino)pentahydrate (C12H26N6O9Zn).
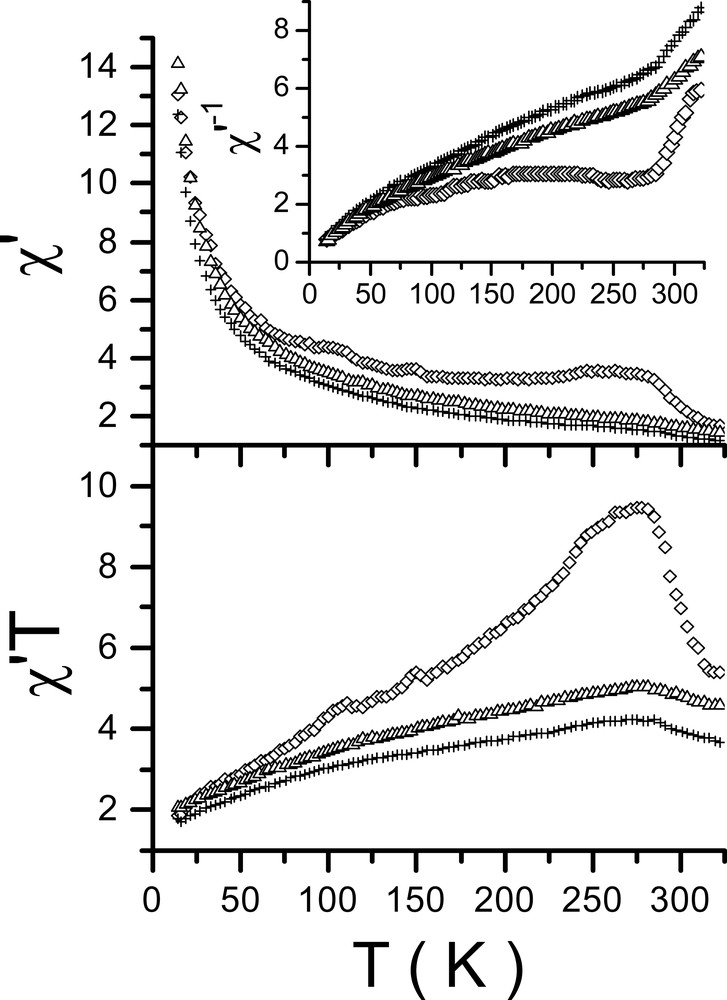
Thermal dependence of χ′ (10–5 emu g–1 Oe), χ′–1 (105 g Oe emu–1) and χ′T product (10–3 emu K g–1 Oe) for 1 (◊), 2 (+) and 3 (△).
3 Conclusions
Two open-framework Nd adipates have been obtained from solutions under ambient conditions. Their AC magnetic susceptibilities indicate a rather complex temperature dependence, which appears to be strongly correlated with the water content and deserves further theoretical and experimental work.
4 Experimental
4.1 Synthesis
Reagents of analytical grade, NdCl3·6 H2O (Aldrich) and adipic acid (Sigma), were used in the synthesis without further purification.
General procedure for compounds 1–3:
Compounds were synthesized by dissolving 0.5 mmol of NdCl3·6 H2O in a 50 ml aqueous solution containing 0.75 mmol of adipic acid under stirring. The final pH 4.5 was reached by addition of NaOH solution.
1- The solution was left to evaporate slowly at room temperature. After crystallization, the product was filtered off, washed with distilled and dried under ambient conditions (65% yield). Prismatic pale violet single crystals, suitable for the crystallographic study were previously collected.
2- The solution was left to evaporate at 80 °C. Prismatic pale violet single crystals, suitable for X-ray diffraction analysis, were collected while the remaining product was filtered off, washed with hot distilled water and dried under ambient conditions (50% yield).
3- After show evaporation at room temperature, the obtained product was filtered off, washed with distilled water and dried under P4O10.
4.2 Single crystal X-ray analysis
Room temperature X-ray diffraction data collections, were performed at 293(2) K using a CAD-4 equipped with a rotating anode generator using graphite-monochromated Mo Kα radiation. The CAD-4 software [31] and the XCAD-4 [32] code were used for data collection and cell refinement and for data reduction, respectively. Low temperature X-ray diffraction data collections were performed at 120(2) K, on a Kappa CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo Kα radiation. Data were collected up to 50° in 2θ, with a redundancy of 4. The final unit cell parameters were based on all reflections. Data collections were made using the COLLECT program [33]; integration and scaling of the reflections were performed with the HKL Denzo–Scalepack system of programs [34]. Absorption corrections were carried out using the multi-scan method [35].
The structures were solved by direct methods with SHELLXS 86 [36] and SHELXS-97 [37]. The models were refined by full-matrix least-squares on F2 with SHELXL-97 [38]. All the hydrogen atoms were stereochemically positioned and refined with the riding model [10]. Hydrogen atoms of the CH2 groups were set isotropic with a thermal parameter 20% greater than the equivalent isotropic displacement parameter of the atom to which each one was bonded. This percentage was set to 50% for the hydrogen atoms of the water molecules. Data collections and experimental details for the complexes are summarized in Table 1. The programs SHELXL97 [38], PLATON [39], PARST [40] and ORTEP-3 [41] were used within WinGX [42] for structure analysis and to prepare materials for publication. Atomic coordinates, bond lengths and angles, and thermal parameters have been deposited by the Cambridge Crystallographic Data Center (see below).
4.3 AC susceptibility measurements
Magnetic measurements were carried out with a LakeShore 7130 AC Susceptometer, with an alternating field of 1 Oe and 825 Hz, in stabilized temperature mode, with a step of 3 K per point, in the range 14–323 K.
5 Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK as CIF files No. CCDC 237654 (1, 120 K), No. CCDC 237655 (2, 120 K), No. CCDC 237657 (1, 298 K), No. CCDC 237656 (2, 293 K) and can be obtained by contacting the CCDC.
Acknowledgments
The authors thank financial support from CONICET (PID 4929 and PIP 0460), ANPCyT (PICT 03041), CNPq (PROSUL: 490172/03), CSIC (Spain), UNLP and UNSL. G.P., G.E., J.C.P. and G.E.N. are members of the CONICET.