

1 Introduction
Organotin hydrides are important reducing agents used in organic synthesis [1]. Like all tin hydrides reported to date, they generally have tin in the formal +4 oxidation state. Until recently there were no stable, well-characterized, divalent tin +2 hydrides, and no equivalent stable hydrides of other group 14 elements had been reported. However, there is now a relatively large body of data showing that low-coordinate, divalent organic derivatives of the group-14 elements can be stabilized by bulky substituents [3]. Reasoning that lower valent hydride derivatives of these elements might be stabilized in an analogous way, Eichler and Power [2] have prepared the first known example of a Sn(II) hydride, [2,6-Trip2C6H3Sn(μ-H)]2·4 C6H6 (1), Trip = 2,4,6-tri-isopropylphenyl, which they have stabilized through the use of the bulky Trip substituent. In view of the novel nature of 1 it was deemed to be important to carry out a neutron diffraction study to locate the hydrides accurately and to give precise Sn–H distances. IR spectroscopy had suggested that, while 1 has a dimeric structure in the solid, it is monomeric in solutions with hydrocarbon solvents. The X-ray structure of 1 at 90 K [2] shows the tin hydride complex to have a dimeric structure with essentially symmetrical hydride bridges and a long Sn–Sn separation, of 3.12 Å, indicative of very little direct tin-tin interaction.
2 Results and discussion
Fig. 1 illustrates the essential features of the molecular structure of the tin hydride complex in 1. To our knowledge this is the first neutron diffraction study of a tin hydride complex to be reported. The dimeric complex in 1 sits on a crystallographic center of inversion assuming a trans geometry with two bridging hydrides. Significantly the two independent Sn–H bonds are found to be of essentially equal length, 1.943(7) Å and 1.941(7) Å, and the effective molecular symmetry of the complex is C2h. The tin atoms carry lone pairs, as has been reported by Eichler and Power [2], and the angles around tin sum to 257° pointing to the pyramidal nature of the coordination at these centers. A listing of important distances and angles in 1 from the neutron diffraction study is included in Table 1. In general, all distances and angles fall within expected limits and are close to their corresponding X-ray values. For an ORTEP [4] drawing of 1, see Fig. 2.
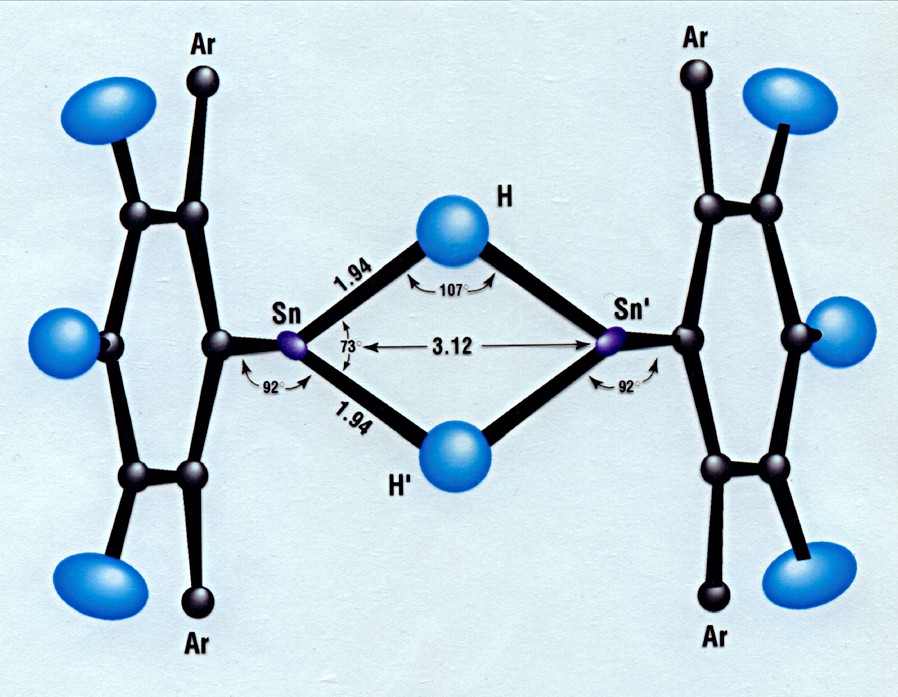
View of 1 along a direction perpendicular to the Sn2H2 plane. The Trip ligand is denoted Ar.
Important distances and angles in 1 from neutron diffraction at 20 K, with the corresponding parameters determined by X-ray diffraction at 90 K [2]
| Distance | Neutron Diffraction | X-ray Diffraction |
| Sn–H | 1.943(7) Å | 1.89(3) Å |
| Sn–H′ | 1.941(7) | 1.95(3) |
| Sn–C | 2.193(4) | 2.210(3) |
| Sn–Sn′ | 3.120(5) | 3.1192(3) |
| H–H′ | 2.31(1) | 2.24(18) |
| Angle | ||
| C–Sn–H | 92.4(2) ° | 91.7(9) ° |
| C–Sn–H′ | 91.6(2) | 93.6(9) |
| Sn–H–Sn′ | 106.9(3) | 108.7(9) |
| H–Sn–H′ | 73.1(3) | 71.3(9) |
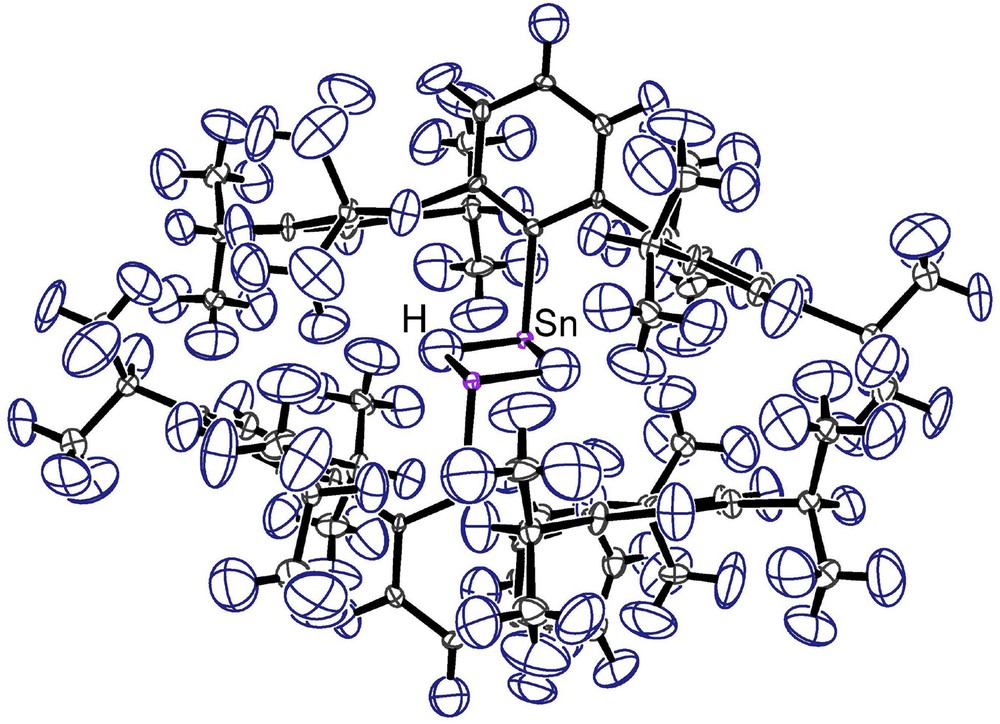
ORTEP [4] diagram of 1 viewed approximately normal to the Sn–C bonds to highlight the pyramidal coordination at the tin atoms. Thermal ellipsoids are plotted at 90% probability.
The neutron diffraction study confirms that 1 contains tin in the +2 oxidation state. Most significantly, no evidence was found in the final difference Fourier synthesis for terminal hydrides on tin.
3 Experimental
3.1 Synthesis and crystal preparation
Crystals of 1 were obtained from a preparation in which a di-isobutylaluminum hydride solution was added drop wise to a solution of 2,6-Trip2C6H3SnCl in diethyl ether at –78 °C. The initially orange solution became green, and subsequently upon warming to ca. 30 °C the color changed to dark blue. After further warming to room temperature, the diethyl ether was removed, and the remaining blue solid was extracted with benzene. Filtration and concentration of the blue solution produced orange crystals of 1.
3.2 Neutron diffraction measurements
A large single crystal of 1, prepared at Davis, was estimated to be of dimensions 1 × 3 × 5 mm, or approximately 15 mm3 in volume. Crystals of 1 are triclinic, P, a = 12.995(1), b = 13.093(1), c = 13.199(1) Å, α = 93.552(8), β = 97.607(8), γ = 115.801(8) °, V = 1986.0(4) Å3 at T = 20 K, Z = 1, with 111 independent atoms.
The crystal was handled in a glove bag under N2 where it was coated with fluorocarbon grease to protect it from contact with air. It was then molded into an aluminum foil pack that was glued to the end of an aluminum pin with DEVCON® 5 minute® epoxy adhesive. The mounted sample was placed in the DISPLEX® closed-cycle refrigerator on the IPNS SCD instrument [5] and was cooled to 20 K.
At IPNS neutrons are produced by a pulsed (30 Hz) spallation source. The SCD time-of-flight Laue instrument, with its time- and position-sensitive detector, accumulates data in x,y,t-histogram form and uses the entire thermal spectrum of neutrons from each pulse. A detailed description of the SCD instrument and data collection and analysis procedures has been published [6,7].
The crystal was indexed based on a preliminary data histogram. The unit cell that was obtained at 20 K approximately matched the 90-K X-ray cell [2], indicating that the neutron crystal was an authentic sample of 1.
Data collection runs of approximately 5–6 h per histogram were initiated arranged at χ and ϕ values suitable to cover a unique hemisphere of reciprocal space (Laue symmetry ). In total 44 histograms were completed during 11 days of measurement on SCD. Altogether, some 27 542 reflections were sampled. Counting statistics were excellent with approximately 100 peaks per histogram observed with I/σ(I) > 10.
The recorded histograms were indexed and integrated using individual orientation matrices for each histogram to allow for any misalignment of the sample. Bragg peaks were integrated in three dimensions about their predicted locations and were corrected for the incident neutron spectrum, detector efficiency, and dead-time loss. A Lorentz correction was also applied. The intensities were corrected for the wavelength-dependent absorption assuming a spherical crystal of radius 1.5 mm.
Structure refinements were carried out with the GSAS system [8], starting with the atomic positions taken from the 90-K X-ray study. All atoms were refined with anisotropic displacement parameters with the exception of four organic hydrogen atoms, for which the displacement parameters tended to become non-positive definite, which were therefore refined isotropically. This model refined satisfactorily, based on the reflections observed above 3σ in intensity, including a Type I Gaussian extinction correction. Outliers for which Fo and Fc differed by a factor greater than 2 were assumed to be affected by systematic errors and were accordingly discarded. Final agreement indices are as follows: Rw(F) = 0.076, GOF = 2.64, for no = 9,932 and nv = 1,024. Extinction was found to be of minimal significance; the maximum correction for strong reflections did not exceed 6%.
A final difference–Fourier synthesis did not show any residual peaks at chemically reasonable positions. In particular, no evidence was found for terminal hydrides on tin. The height of the largest positive residual peak was observed to be approximately 4% of a typical carbon Fourier peak while the height of the largest negative residual peak was approximately 15% of a typical hydrogen Fourier peak.
Acknowledgements
We would like to thank Ms. Martha Miller for assistance with the neutron diffraction experiment. We are also indebted to Dr. Vincent Leon for translating our abstract into French. Work at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract W-31-109-ENG-38. NATO provided additional financial support under grant PST.CLG.976225.