

1 Introduction
1.1 Scope of the article
The defined pore structures of molecular sieves are ideal for obtaining organized arrangements of a large variety of guest species [1]. In order to fully exploit the potential of these host–guest systems it is essential to control the interaction of a given molecular sieve particle with its environment. This article discusses methods for the functionalization of the external crystal surface of zeolites; thereby focusing on our research on zeolite L. Concepts are presented which allow for assigning functionalities to the whole external surface or selectively to the regions of the external surface where the channel entrances are located.
1.2 Zeolite L
Zeolite L is a crystalline aluminosilicate featuring hexagonal symmetry [2–4]. The crystals consist of cancrinite cages linked by double six-membered rings, thereby forming columns in the c-direction. Connection of these columns gives rise to 12-membered rings with a free diameter of 7.1 Å. As a consequence, zeolite L possesses one-dimensional channels running parallel to the c-direction through the entire crystal. The free diameter varies from 7.1 Å (narrowest part) to 12.6 Å (widest part). The main channels are linked via non planar eight-membered rings forming an additional two-dimensional channel system with openings of about 1.5 Å. As can be seen from the scanning electron microscopy image shown in Fig. 1, the morphology of the crystals can be approximated by a cylinder, with the entrances of the main channels located at the base. The number of channels is equal to 0.265(dc)2, where dc is the diameter of the cylinder in nanometers. A crystal with a diameter of 550 nm, therefore, consists of about 8 × 104 parallel channels.
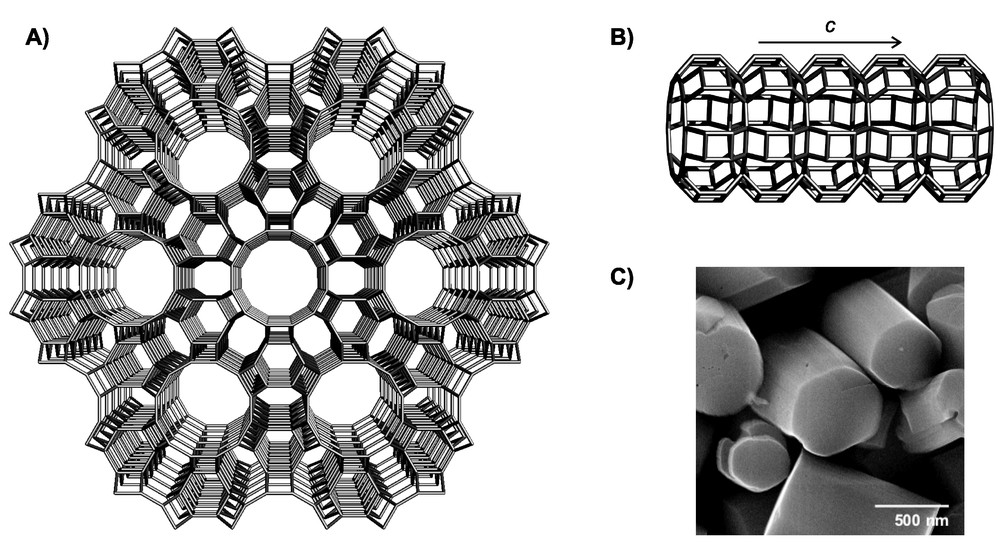
Framework and morphology of zeolite L. (A) Framework viewed along the c axis, (B) side view of the main channel, (C) scanning electron microscopy image of zeolite L crystals.
The substitution of Si4+ by Al3+ creates an anionic framework. Charge compensation is accomplished by additional cations (usually Na+ and K+). The stoichiometry of zeolite L containing monovalent cations M is M9[Al9Si27O72]·nH2O, where n is 21 in the fully hydrated state, and 16 at about 20% relative humidity. The water molecules in the large cavities of zeolite L have been reported to behave like an intracrystalline liquid, whereas they seem to build clusters around the cations in the smaller pores [5].
1.3 Dye–zeolite composites
The inclusion of cationic or neutral dye molecules into the one-dimensional channels of zeolite L yields highly organized composites exhibiting a variety of intriguing properties which we have extensively reviewed in Refs. [1,6,7]. The efficient transport of electronic excitation energy by the chains of electronically non-interacting chromophores is the most prominent feature of this material. Fig. 2 illustrates a bidirectional photonic antenna based on this principle. Light is absorbed by the strongly luminescent dye molecules in the channels. Due to the short distances and the ordering of the electronic transition dipole moments, the electronic excitation energy can be transported by Förster energy transfer to a specific trap (acceptor) located at the channel ends. Transport of the excitation energy is radiationless and occurs preferentially along the channel axis. For a detailed theoretical account of Förster energy transfer in dye-loaded zeolite L see Ref. [8].
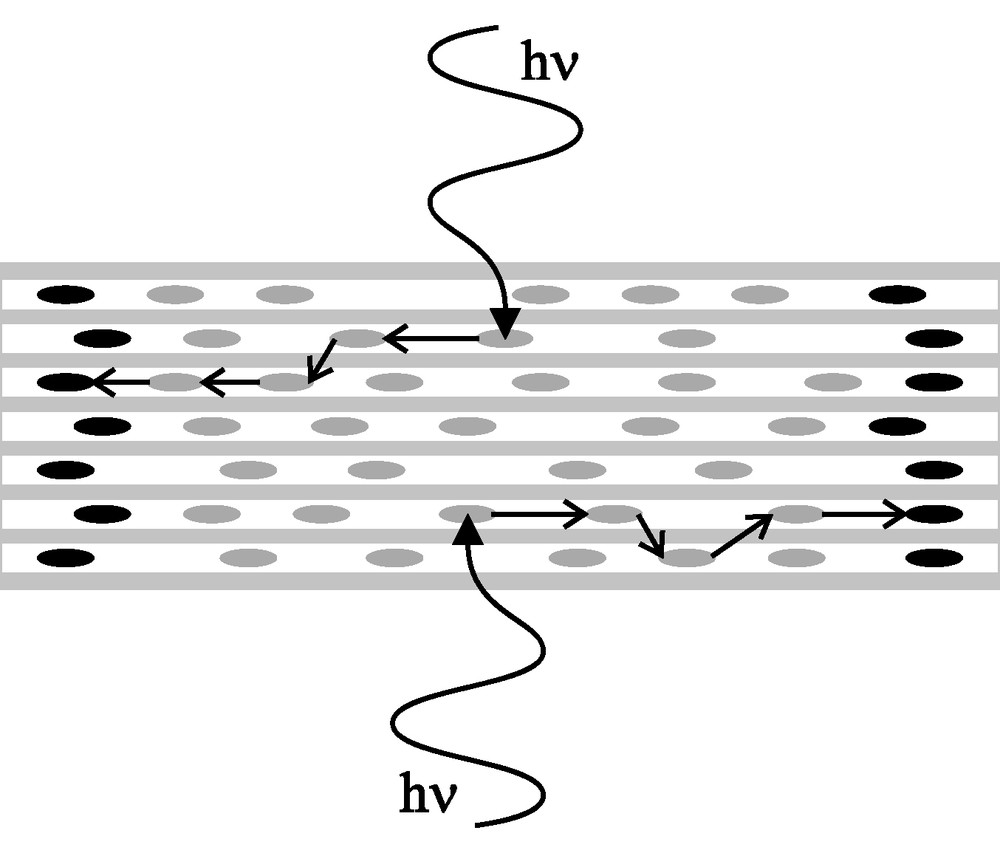
Schematic representation of a bidirectional photonic antenna based on zeolite L containing donor dyes (grey) and acceptor dyes (black).
Control of the morphology and size of the host crystals allows optimization of the dye–zeolite composites for a specific application. Large crystals (>2 μm) are useful for studying the photophysical properties by optical microscopy while small crystals are ideal hosts for the preparation of high efficiency photonic antenna systems. Disc-shaped crystals are used to obtain oriented monolayers of zeolite L. The functionality of the dye-loaded zeolite crystals can be extended by coupling them to external devices [9]. The ability to do so implies controlling the external surface properties of the zeolite.
2 Functionalization of the external surface of zeolite L
2.1 Basic principles
Surface hydroxyl groups are convenient targets for modification. Besides the catalytically important bridging Si(OH)Al groups (Brønsted acid sites), zeolites also contain SiOH groups (silanols). Using IR spectroscopic probes and zeolite ZSM-5 in its protonic form, Otero Areán et al. [10] found that Brønsted acid sites are generally located on the internal surface while silanol groups are the dominant species on the external surface where they saturate dangling bonds created by truncation of the framework. In principle, silanols should not be present in the channels or cavities of perfect crystals. Silanols located on the internal surface of zeolites are usually associated with defect sites formed for example by dealumination [11].
In the majority of cases, functionalization of the external surface of zeolites is achieved by reacting the surface silanols with organosilanes of the type RnSiX(4–n). X is the reactive group (typically halogen or alkoxy) and R is a nonhydrolyzable moiety possessing the desired functionality. Zhan et al. [12] modified the external surface of nanometer-sized zeolite X crystals with amino groups (by reaction with 3-aminopropyltriethoxysilane, APTS) and vinyl groups (by reaction with vinyltriethoxysilane, VTS). Based on Raman and solid-state NMR spectra it was proposed that reaction with APTS leads to a dipodal grafting RSi(OSizeolite)2, whereas tripodal grafting RSi(OSizeolite)3 is obtained in the case of VTS. The difference in podality between the two modifications was attributed to the different hydrolytic activity of the organosilane reagents. The presence of reactive surface-anchored groups opens possibilities for further functionalization. Surface-anchored amino groups are frequently used for this purpose. Reactions of zeolite-grafted amino groups with isothiocyanates [13], chlorides [12,14], epoxides [15], and fullerenes [16] have been reported.
Studies on the adsorption of APTS on silicon oxides have shown that the mechanisms leading to the formation of siloxane bonds are complex and depend on several factors, such as hydration of the surface, reaction temperature and duration, and curing treatment. These parameters also influence the extent of covalent cross-linking between the APTS molecules [17]. As an alternative to organosilanes, isocyanates can be employed for surface modification. 1,4-Diisocyanatobutane for example was observed to readily form urethane linkages with the surface hydroxyl groups of various zeolites [18].
Modifications following the above principles usually affect the entire external surface of the zeolite crystals. This can be demonstrated by a simple experiment: After reaction of zeolite L with an excess of APTS in dry toluene (room temperature), the surface-anchored amino groups were coupled to a fluorescence marker (fluorescein isothiocyanate FITC). Examination of single zeolite crystals under a fluorescence microscope revealed a uniform coverage of the external surface (see Fig. 3). An efficient method to detect the surface-bound amino groups is by reaction with ninhydrin, which is known to be readily converted to Ruhemann’s Purple (ɛ = 22,000 at λmax = 570 nm) upon reaction with various alkylamines [16,19].

Fluorescence microscopy image of zeolite L with fluorescein covalently bound to the external surface. The length of the cylindrically shaped crystals is approximately 5 μm. The crystal on the right is standing on its base.
The reactivity of alkoxysilanes towards surface hydroxyls strongly depends on the solvent. Room temperature reaction of an excess of APTS with zeolite L was found to produce multilayer coverages in solvents of low polarity (toluene, hexane). Coverage decreased with increasing polarity, i.e. reaction in THF or CH2Cl2 produced sub-monolayer coverages. The result of a corresponding ninhydrin assay is shown in Fig. 4.

Ninhydrin assay of zeolite L after room temperature modification with APTS in toluene (solid line) and APTS in THF (dashed line). Samples were repeatedly washed with ethanol after the modification in order to remove physisorbed APTS. The band at 360 nm is due to unreacted ninhydrin. The product (Ruhemann’s purple) bands at 405 and 570 nm indicate a significantly larger amount of surface-bound amino groups when toluene is used as a solvent.
There are only few reports on the functionalization of specific parts of the external zeolite surface: by reacting octadecyltrichlorosilane with aggregated zeolite Y particles and making use of the fact that only the exposed external surfaces of the aggregates are accessible, Ikeda et al. [13] achieved partial modification. The resulting particles of zeolite Y featured both hydroxyl and alkylsilyl faces, thus exhibiting amphiphilic properties. In the case of zeolite L, one can make use of the specific interaction of molecules with the channel entrances at the base planes of the cylindrical crystals to accomplish a highly selective partial functionalization of the external surface. This principle, which we have termed Stopcock Principle, will be discussed in Section 2.2.
Discriminating between the internal and external surface of zeolites is not always trivial. As zeolites belong to the class of microporous materials (IUPAC classification [20]) and thus feature pore widths smaller than 2 nm, specific modification or probing of the external surface is often successful by choosing molecules which are too large to enter the pores [21]. Additionally, hydroxyl groups on the external surface are more exposed than those in the channels and are therefore the most kinetically available. This latter fact allows the discrimination between external and internal surface even in the case of mesoporous silica (pore width ranging from 2 to 50 nm), where dichlorodiphenylsilane is usually applied to passivate the external surface before directed derivatization of sites in the pores is conducted [22].
Cationic dyes, which we frequently use as guest molecules [1], have the tendency to form aggregates on the external surface of zeolite L. The distinctively different photophysical properties of these aggregates compared to the corresponding monomers in the channels (i.e. adsorbed on the internal surface) can be used to distinguish between the two cases. We have also applied solvatochromy to check whether molecules are located on the external surface. In this method, the dye–zeolite composite is suspended in a solvent, which for reasons of polarity or sterical hindrance cannot enter the zeolite channels. Molecules on the external surface will then experience a different environment resulting in a change of the electronic absorption spectrum depending on the solvent [7].
2.2 Selective modification at the channel entrances (Stopcocks)
Typical stopcock molecules are composed of three parts: a narrow label which can penetrate into the channels of the zeolite, a head too large to enter the channels, and a spacer connecting head and label (see Fig. 5). We will in the following frequently refer to label and spacer as the tail of a stopcock molecule. While the head moiety carries the desired functionality for the external surface, the label and/or spacer should provide polar groups, such as carbonyl or nitro groups, exhibiting a favorable interaction with the internal surface of the zeolite [23,24]. Spacers should be flexible to have the capability to adapt to the channel entrances (e.g. aliphatic chains, polyethers, amides). We distinguish between reversible stopcocks [25,26] attached to the channel entrances by adsorption and irreversible ones anchored by covalent bonds. An example for each type is shown in Figs. 5 and 6. Both molecules are coumarin derivatives with the coumarin moiety featuring as the label. An irreversible stopcock (see Fig. 6) has to provide groups, which can be reacted with the surface hydroxyl groups at the channel entrances after completion of the adsorption process. Alkoxysilyl groups have proven to be well suited for this purpose. The solvent from which the stopcock is adsorbed onto the zeolite has to be chosen so that the solubility of the stopcock is low while inhibiting reaction of the alkoxysilyl group with the surface hydroxyls. The latter assures that the stopcocks are oriented correctly (tail inside the channel) before covalent anchoring takes place. In the case of the triethoxysilylated coumarin depicted in Fig. 6, adsorption, i.e. reversible insertion of the coumarin moiety into the channels, was carried out from CH2Cl2. After centrifugation and drying in a stream of N2 the resulting material was refluxed in toluene to allow the formation of siloxane bonds [27]. It is very likely that the number of siloxane bonds between stopcock and zeolite is restricted to two. The remaining ethoxy group, therefore, corresponds to the functionality A, which could be used for a further coupling reaction.
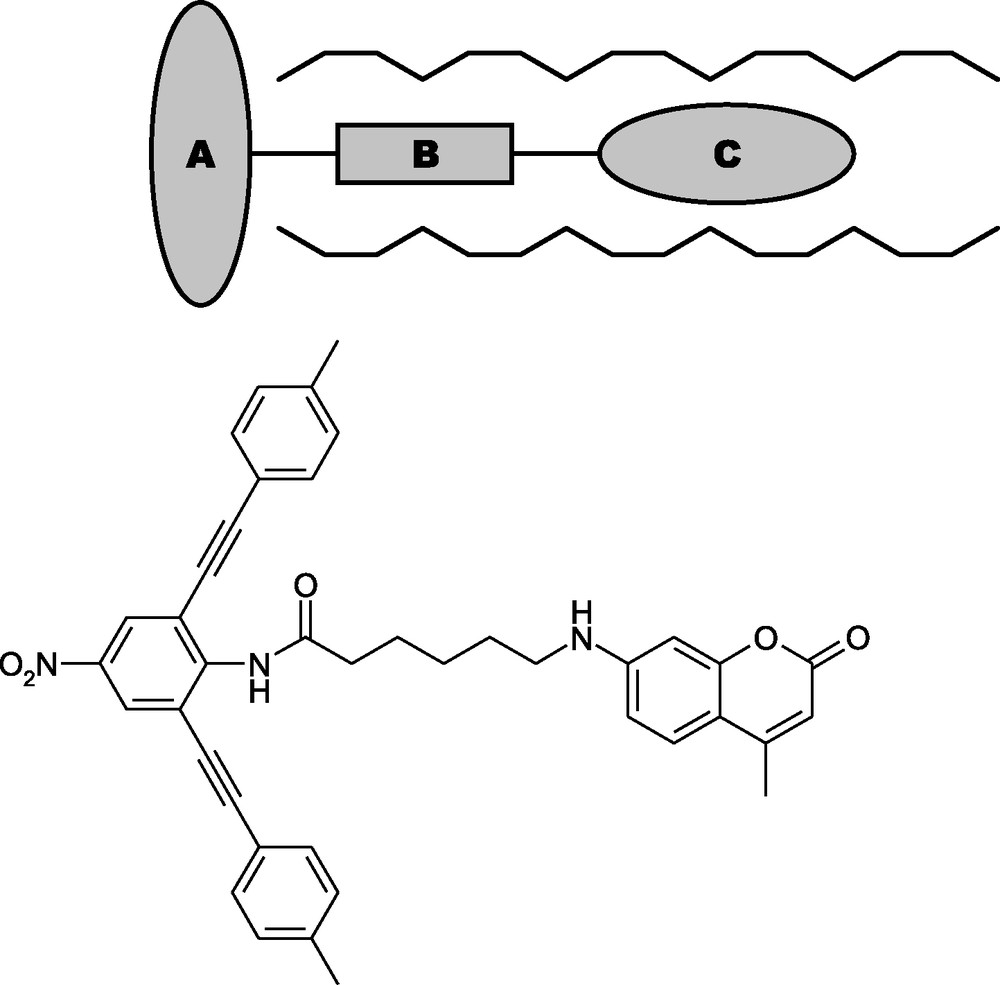
Schematic representation and example of a reversible stopcock consisting of head (A), spacer (B), and label (C) moieties.

Schematic representation of an irreversible stopcock consisting of head (A), spacer (B), label (C), and an alkoxysilyl group for covalent bonding to the channel entrance. The molecule shown as an example contains a triethoxysilyl group to accomplish covalent anchoring.
The location of stopcocks containing a fluorescent moiety can be determined by examination of single zeolite crystals under a fluorescence microscope. As can be immediately seen from the images shown in Fig. 7, the fluorescent stopcock molecule BTRX is adsorbed selectively at the base (channel entrances) of the cylindrical zeolite crystals. Upon consideration of the polarization of the fluorescence it becomes evident that the BTRX emission is polarized perpendicular to the zeolite channel axis. Since the S0 ← S1 transition dipole moment of the BTRX chromophores is mainly polarized perpendicular to its tail [28], we can conclude that the tail of the stopcock has penetrated into the channel while the head remains outside [25].
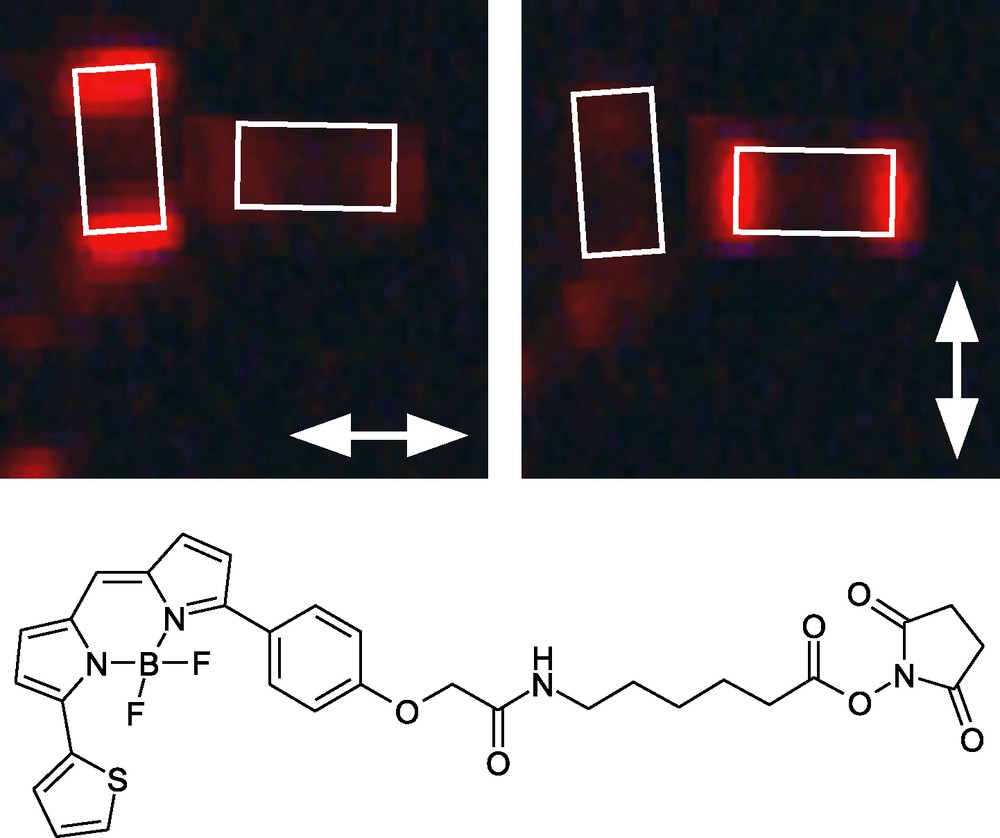
Fluorescence microscopy images of zeolite L modified with BTRX [25]. The white rectangles indicate the position of the zeolite crystals (approximately 2 μm in length). The transmission direction of the polarizer is given by the arrows.
The Stopcock Principle allows tuning of the external surface properties by varying the head moieties. BTRX for example is used for extracting electronic excitation energy from dye–zeolite composites [25]. Light absorbed by donor molecules located in the channels is transported through migration of the electronic excitation energy. Due to a spectral overlap between the donor emission and the stopcock head absorption, energy transfer can occur, thus trapping the electronic excitation energy at the external surface. The system can be reversed by filling the channels of the zeolite with acceptor molecules and employing a stopcock head with donor properties. This enables energy injection from the stopcock head located at the external surface to the acceptor molecules in the zeolite channels. Head moiety functions other than energy transfer are conceivable. We are currently investigating stopcock molecules with fullerenes as head moieties. Being efficient electron acceptors [29,30], such stopcock heads open possibilities for the investigation of electron transfer processes in zeolites [6].
3 Coupling dye–zeolite composites to external devices
The external surface of a zeolite constitutes the interface through which communication between guest molecules in the channels or cavities and species on the outside occurs. Functionalization of the external surface provides a tool for tuning this interface to efficiently perform a certain task (e.g., energy transfer) or exhibit a desired property (e.g., hydrophobicity). Besides tuning the interface by functionalization of the external surface, control of size and morphology of the zeolite crystals is important in order to accomplish ideal interaction between zeolite and external device. A forthcoming publication will describe methods to tune the size (30–7000 nm) and the morphology (rod-shaped to disc-shaped) of zeolite L crystals.
Embedding of dye-loaded zeolite crystals into polymers is of interest for various reasons. The coupling of dye–zeolite photonic antennae to a luminescent or photoconductive polymer opens possibilities for the development of novel LED materials. Energy transfer from the polymer to the zeolite or vice versa is possible via suitable stopcock molecules located at the channel entrances. The energy transfer can be enhanced by covering the external surface of the zeolite with acceptor or injector molecules. This is accomplished by reaction with APTS and subsequent coupling of the zeolite-bound amino groups to an amino-reactive fluorescent dye. We have recently prepared a polymer-embedded dye–zeolite L material, for which we observed transfer of electronic excitation energy from the polymer to the dye molecules at the external zeolite surface and ultimately to the dye molecules located in the zeolite channels [31]. A large variety of dyes covering the whole visible spectrum can be included into zeolite L [1]. Embedding small crystals (<50 nm) of dye-loaded zeolites into polymers hence allows color tuning without the need of adapting the procedure of polymer preparation.
Stopcock molecules can be used to mediate the electronic excitation energy transfer from a dye-loaded zeolite (=photonic antenna) to a semiconductor. In a recently investigated system, radiationless excitation energy transfer from a dye inside the channels of zeolite L to a dye covalently bound at the external surface and further through a thin insulating layer of SiO2 (preventing electron transfer) to a silicon semiconductor was observed [32].
4 Conclusions
Zeolites are ideal host materials for supramolecular organization. Besides their well-defined pore structure, they also feature silanol groups on the external surface. The presence of these reactive sites opens possibilities to functionalize the external surface and thus allows tuning of the interface between the guest molecules in the channels/cavities and the environment. By exploiting the interaction of molecules with the pore openings of a zeolite, selective functionalization becomes possible in cases where the openings are located on specific parts of the crystal surface. This Stopcock Principle can also be used to close the pore openings of a zeolite in order to inhibit leaching of the guests or to provide additional protection of the guests from external species.
Acknowledgements
Financial support by the Swiss National Science Foundation (Project NFP 47, 4047-057481) is acknowledged. The authors also thank the Bundesamt für Bildung und Wissenschaft (BBW Switzerland) for financial support within the European Union Project ‘RTN Nanochannel’. The experiments reported in Figs. 3 and 4 were conducted by Quirin Scheifele. Framework images shown in Fig. 1 were prepared by Dr. Antonio Currao.