

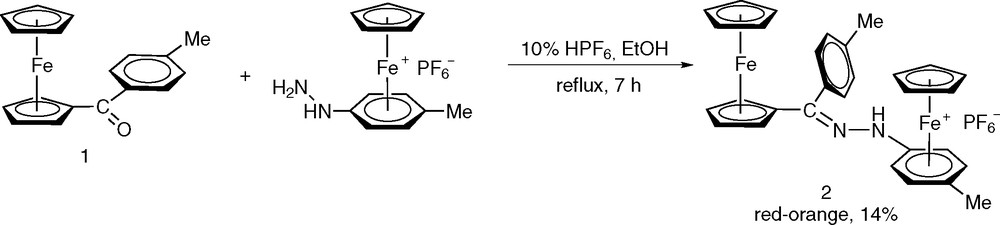
1 Introduction
The molecular sciences of the transition metals have potential applications in the new fascinating field of molecular electronics [1–3], based inter alia on the fact that the oxidation states of transition metals can be varied to a great extent and thus that many electron transfer processes can result. By suitable molecular engineering, it should become possible to assemble and tune molecular devices including transition metals and organize their interface with the macroscopic world. In this context, we have focused our attention on simple bimetallic organoiron(II) model systems [4–7], made of the cationic [CpFe(η6-arene)]+ (Cp = η5-C5H5) moiety as an electron withdrawing building block [8–10] linked to a donating ferrocenyl unit [11] through the hydrazonic skeleton spacer, –NH–N=CR– (R = H, CH3), in order to investigate the electronic cooperation between both metal centers. Structural, electronic and theoretical (DFT) data have confirmed the ground state electronic interaction between the electron withdrawing mixed sandwich [CpFe(η6-arene)]+ and the donor ferrocene-based termini [4]. In the pursuit of our previous work, we have investigated the stereo-electronic effects of the ketone substituents on the condensation reaction, and the present contribution describe (i) the preparation and full characterization (IR, 1H NMR, UV–vis, cyclic voltammetry) of a new Type I non-rod-shaped dipolar chromophore [12] formulated as [CpFe(η6-p-CH3C6H4)–NHN=C(C6H4-p-CH3)(η5-C5H4)FeCp]+PF6–, (ii) its X-ray crystal structure showing the unexpected syn-conformation of the two organometallic moieties, and (iii) the crystal structure of the toluoylferrocene precursor.
2 Results and discussion
2.1 Synthesis and characterization
The preparation of the bimetallic hydrazone complex [CpFe(η6-p-CH3C6H4)NHN=C(C6H4-p-CH3)(η5-C5H4)FeCp]+PF6– (2) was achieved by reaction of the ionic organometallic hydrazine precursors [CpFe(η6-p-CH3C6H4–NHNH2)]+PF6– with 1 equiv of the sterically hindered toluoylferrocene CpFe(η5-C5H4)CO(C6H4-p-CH3) (1), in ethanol solution containing 10% of hexafluorophosphoric acid, HPF6, as catalyst (Scheme 1). The suspension was refluxed for 7 h under dinitrogen. Complex 2 was isolated as an air stable orange solid in 14% yield. All other attempts to increase the yield and to synthesize such bimetallic hydrazones starting from the same hydrazine precursor and the ferrocene-based ketones, CpFe(η5-C5H4)CO(p-RC6H4) (R = H, F) or the diferrocenylketone, failed, whatever the acid catalyst used (glacial acetic acid, HPF6). The low reactivity or reluctance of the above mentioned conjugated ketones to undergo condensation reaction with hydrazines can be ascribed to the delocalization of the positive charge density on the aromatic rings as indicated by the low infrared stretching vibration of the carbonyl group at 1628 cm–1 for 1 [13], and to a lesser extent, to the steric bulk around the carbonyl carbon [14].
Compound 2 exhibits a good solubility in common polar organic solvents, but is insoluble in diethyl ether, hydrocarbons and water. Its solid IR spectrum exhibits the characteristic features usually encountered for such bimetallic hydrazones [4]. Those are (i) a weak absorption band at 3289 cm–1 attributed to the ν(N–H) stretching vibration; (ii) a medium band at 1559 cm–1 assigned to the ν(C=N) stretching mode, (iii) a very strong ν(PF6) bands at 825 cm–1, and a strong δ(P–F) band at 554 cm–1.
The unique set of signals observed in the 1H NMR spectrum (acetone-d6, see Section 4.2.) clearly indicate the stereoselective formation of compound 2, as the sterically less hindered trans-isomer about the N=C double bond. This is in agreement with previous NMR studies of related derivatives [CpFe(η6-p-CH3C6H4)NHN=CR-(η5-C5H4)FeCp]+PF6– (R = H, 3; CH3, 4) [4], and definitively assigned from its crystal structure (see below). Interestingly, the benzylic N–H resonance is identical to that found for 4 (δ = 8.71). This signal is upfield shifted by 0.88 ppm with respect to that observed for 3 (δ = 9.49), suggesting that this is a pure electronic effect.
The UV–visible spectrum of compound 2, in CH2Cl2, is very similar to those of 3 and 4 and analogous bimetallic hydrazones [4–7], and is consistent with most ferrocenyl chromophores in that they exhibit two charge-transfer bands in the visible region [15]. The prominent band at 323 nm is assigned to a ligand-centered π–π* electronic transition, and the less energetic and weaker band at 454 nm responsible for the bright orange color of this compound, is attributed to a metal-to-ligand charge-transfer (MLCT) process (see Section 4.2.). This assignment is in accordance with the latest theoretical treatment (model III) reported by Barlow et al. [16]. The bands associated with the d–d transitions of the cationic mixed sandwich [17,18] and of the ferrocenyl [19] entities are masked by the MLCT band. A very weak solvatochromic effect is observed when the spectrum is recorded in DMSO (see Section 4.2.) [7].
2.2 Electrochemical studies
An electrochemical study in acetonitrile was carried out at room temperature (see Section 4.1. for experimental details). The cyclic voltammetric (CV) response of the homobimetallic hydrazone 2 shows a reversible oxidation process at +0.090 V, and an irreversible reduction wave at –1.990 V (internal reference Cp2Fe/Cp2Fe+), a behavior similar to that of its closely related derivatives 3 and 4 (Table 1). The reversible one-electron oxidation process arises from the oxidation of the monosubstituted ferrocene unit [20], and corresponds to the generation of the dicationic Fe(II)/Fe(III) mixed valence species. On the other hand, the irreversible cathodic process is centered at the mixed sandwich moiety, and is associated to the single-electron reduction of the d6, Fe(II), 18-electron complexes to the unstable d7, 19-electron Fe(I) species [18,21]. Thus, it is clear that in complex 2 the nature of the HOMO is still dominated by the neutral donating ferrocenyl units, whereas the character of the LUMO is determined by the cationic mixed sandwich, in accordance with previous experimental and theoretical work [4].
Comparative electrochemical data for compounds [CpFe(η6-p-CH3C6H4)NHN=C(R)(η5-C5H4)FeCp]+PF6– a
| Compound | Epc, CpFe+(arene) based b | E1/2 (ΔEp/mV) ferrocene-based b | References |
| 2 (R = p-CH3C6H4) | –1.99 | +0.090 (70) | This work |
| 3 (R = H) | –1.90 | +0.130 (89) | [4] |
| 4 (R = CH3) | –2.03 | +0.090 (76) | [4] |
a Recorded in acetonitrile at 298 K with a vitreous carbon working electrode, 0.1 M n-Bu4N+PF6– as supporting electrolyte, scan rate = 0.1 V s–1.
b Potential values in V with reference to ferrocene/ferrocenium couple under the same conditions.
The E0 values of the reversible anodic process is 90 mV anodically shifted with respect to ferrocene, indicating some degree of electronic interaction between the iron centers. Interestingly, this anodic shift is the same as that measured for compound 4, corroborating the imine carbon substituent electronic effect observed in 1H NMR (see above). This donating ability pattern (CH3 > p-C6H4CH3 > H) is also noted for the reduction potentials (Table 1).
2.3 X-ray crystallographic studies
2.3.1 Crystal and molecular structure of 1
Single crystals of p-toluoylferrocene CpFe(η5-C5H4)CO(C6H4-p-CH3) (1), suitable for X-ray structure determination, were grown at –30 °C by diffusion of hexane into a saturated CH2Cl2 solution of 1. Data from the structural study are presented in Section 4.3., important bond lengths and angles are depicted in Table 2, and an ORTEP view of the molecular structure of 1 with the atom labeling scheme is presented in Fig. 1. Complex 1 consists of a neutral mononuclear Fe(II) species. The individual molecules of CpFe(η5-C5H4)CO(C6H4-p-CH3) are well separated from each other with essentially no intermolecular contact distances of less than the sum of van der Waals radii. Compound 1 crystallizes in the orthorhombic space group P212121 with four molecules in the unit cell.
Bond distances (Å) and angles (°) for CpFe(η5-C5H4)CO(C6H4-p-CH3) (1)
| Bond distances | |||
| Fe(1)–C(6) | 2.028(7) | C(6)–C(11) | 1.435(10) |
| Fe(1)–C(1–5)av. | 2.041 | C(11)–O(1) | 1.223(9) |
| Fe(1)–C(6–10)av. | 2.037 | C(11)–C(12) | 1.522(11) |
| Angles | |||
| C(6)–C(11)–O(1) | 121.3(7) | C(6)–C(11)–C(12) | 120.4(6) |
| C(12)–C(11)–O(1) | 118.3(6) |
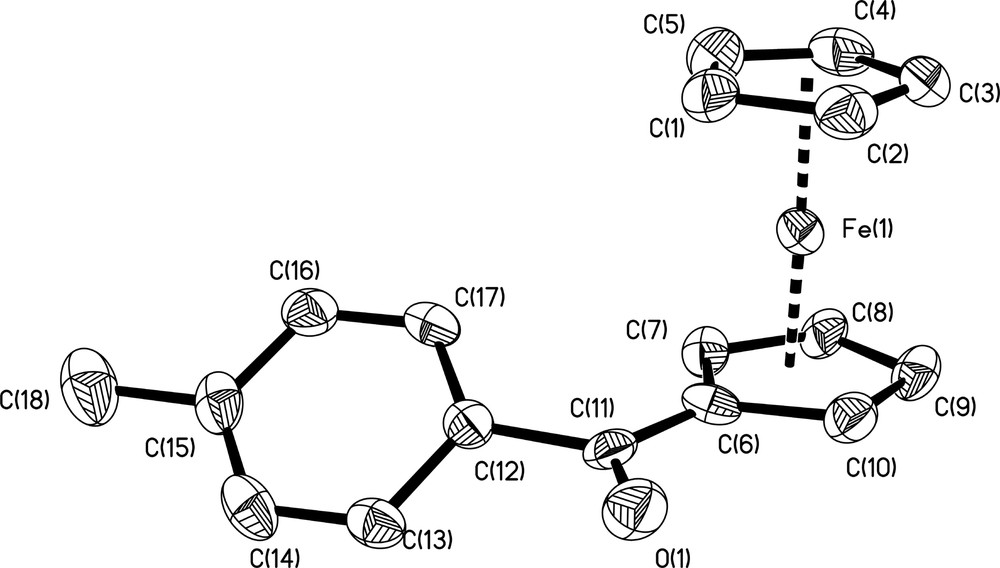
Molecular structure and atom numbering scheme for CpFe(η5-C5H4)CO(C6H4-p-CH3) (1). Hydrogen atoms have been omitted for clarity. Displacement ellipsoids are at the 35% probability level.
The molecular structure of 1 reveals almost eclipsed cyclopentadienyl rings for ferrocene. The iron atom is coordinated to the cyclopentadienyl rings at a ring centroid–iron distances of 1.637 and 1.654 Å for the substituted and the unsubstituted ring, respectively. The two C5-ligands are essentially parallel, with the ring centroid–iron–ring centroid angle of 177.2°. The dihedral angle between the substituted C5-ring and the plane of the tolyl group is of 36.8°, slightly weaker than the 38.3, 38.4 and 42.3° values found for the three other structurally characterized aroylferrocenes, CpFe(η5-C5H4)CO(C6H4-p-R) reported in the Cambridge structural data base [22] where R = H [23], OH and NH2 [24]. The major difference comes from the dihedral angle between the same C5-ring and the C=O axis which is of 3.8° for 1, whereas it is of 14.7, 15.7 and 17.9° for the p-H, p-OH and p-NH2 substituted derivatives, respectively [22,23]. No reason is obvious for this dichotomy of behavior.
2.3.2 Crystal and molecular structure of 2
Single crystals of [CpFe(η6-p-CH3C6H4)NHN=C(C6H4-p-CH3)(η5-C5H4) FeCp]+PF6– (2) suitable for X-ray analysis were grown from a CH2Cl2/diethyl ether solution at room temperature. Data from the structural study are presented in Section 4.3., selected bond lengths for the cationic organometallic unit are presented in Table 3. An ORTEP view of the cation, with atom numbering, is shown in Fig. 2. Compound 2 crystallizes in the triclinic space group P
Bond distances (Å) and angles (°) for [CpFe(η6-p-CH3C6H4)NHN=C(C6H4-p-CH3)(η5-C5H4)FeCp]+PF6– (2)
| Bond distances | |||
| Fe(1)–C(6) | 2.165(7) | C(6)–N(1) | 1.366(9) |
| Fe(1)–C(7–12)av. | 2.072 | N(1)–N(2) | 1.354(8) |
| Fe(1)–C(1–5)av. | 2.005 | N(2)–C(13) | 1.294(8) |
| Fe(2)–C(21) | 2.042(7) | C(13)–C(21) | 1.457(10) |
| Fe(2)–C(21–25)av. | 2.047 | Fe(2)–C(26–30)av. | 2.035 |
| Angles | |||
| C(6)–N(1)–N(2) | 118.3(6) | N(1)–N(2)–C(13) | 117.8(6) |
| N(2)–C(13)–C(14) | 124.5(7) | N(2)–C(13)–C(21) | 117.8(7) |
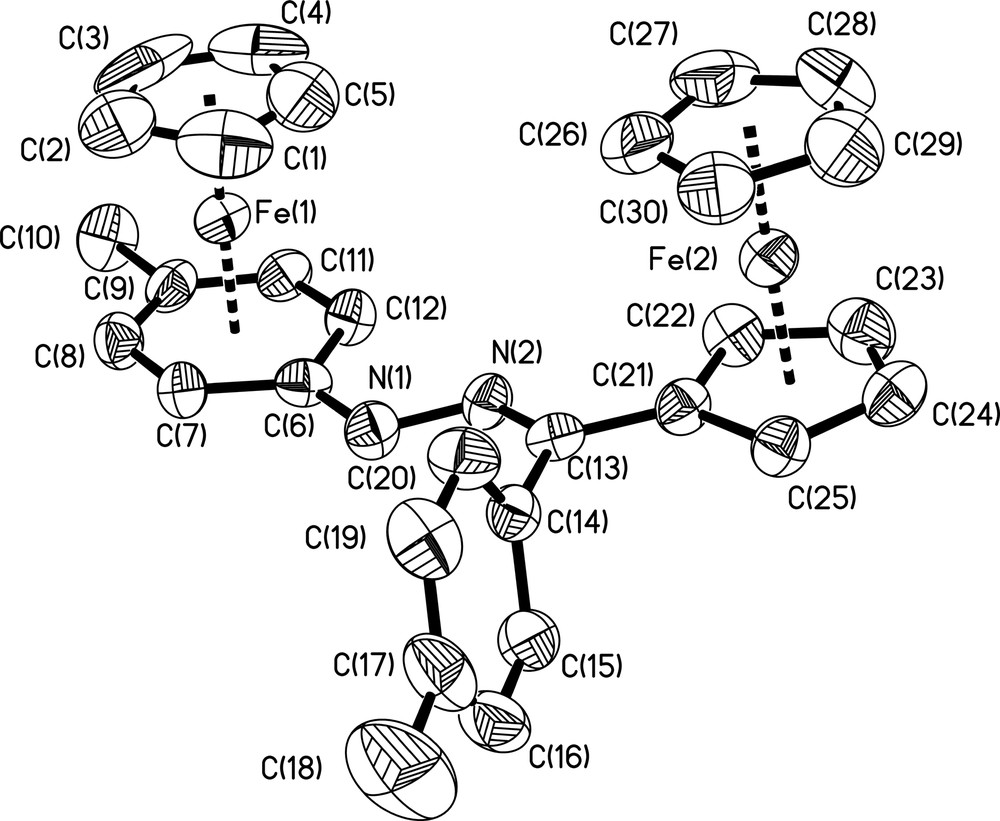
Molecular structure and atom numbering scheme for the cation [CpFe(η6-p-CH3C6H4)NHN=C(C6H4-p-CH3)(η5-C5H4)FeCp]+ (2+). Hydrogen atoms and counter anion PF6– have been omitted for clarity. Displacement ellipsoids are at the 35% probability level.
Complex 2+ adopts the unexpected apparently more crowded syn-conformation [4] with the two iron atoms on the same faces of the anionic π-dinucleating hydrazonyl ligand [p-CH3C6H4–NHN=C(C6H4-p-CH3)–C5H4]– (Fig. 2), with a through-bond Fe···Fe distances of 9.676 Å, and a through-space Fe···Fe distance of 6.791 Å. It is noteworthy that the steric requirement of the p-tolyl substituent at the imine carbon atom and/or packing forces must induce the formation of the syn-isomer in the solid state, whereas only the anti-isomer is found when the hydrazonyl spacer is unsubstituted [25], and that both syn- and anti-rotamers are present in the asymmetric unit of compound 4 bearing a methyl substituent at the imine carbon atom [4]. Theoretical investigations for compound 4 have shown that the anti-isomer is more stable than the syn-isomer by 0.12 eV [4]. On the other hand, the plane of the p-tolyl substituent at C(13) is now almost perpendicular to the substituted cyclopentadienyl ring of the ferrocenyl fragment (dihedral angle = 82.1°), thus illustrating the steric bulk at imine carbone upon formation of the complex. Interestingly, the coordinated C6- and C5-rings of the hydrazonyl spacer are almost coplanar (dihedral angle = 8.1°) allowing an efficient π-electron delocalization or electronic interaction between the electron-donating and electron-accepting termini through the entire hydrazonato skeleton. This is also indicated through the three characteristic structural features observed in such homobimetallic hydrazone compounds [4,5,7,25]: (i) the slight cyclohexadienyl-like character at the coordinated C6 phenyl ring with a long Fe(1)–C(6) bond distance of 2.165(7) Å, and a folding angle of 6.1°, (ii) some Cipso–N multiple bond character as indicated by the C(6)–N(1) distance of 1.366(9) Å, and (iii) the sp2-hybridization of the benzylic nitrogen atom, which is reflected by a bond angle C(6)–N(1)–N(2) of 118.3(6)°.
3 Conclusion
To sum up, we have shown the limitation of the synthesis of organodiiron(II) complexes containing a short π-conjugated hydrazonyl spacer linking the cationic electron-acceptor mixed sandwich, [CpFe(η6-arene)–]+, to the sterically hindered electron-donating ferrocenyl carbene fragment [CpFe(η5-C5H4)CR] (R = p-CH3C6H4 (2), H (3), CH3 (4)). The role of the steric demand of the R substituent at the imine carbon atom is nicely illustrated in the solid state. The anti-isomer is exclusively formed for the unsubstituted derivative (R = H), both anti and syn-isomers are observed for R = CH3, and only the syn-isomer is present when R = p-CH3C6H4. Based on 1H NMR and electrochemical data, both the p-tolyl and the methyl imine carbon substituents exert the same electronic influence. On the other hand, the homobimetallic compound 2 can be designed, from a structural point of view, as Type I non-rod-shaped dipolar chromophores [12], and the data obtained from the UV–vis spectrum and cyclic voltammetry demonstrate electronic delocalization along the entire π-conjugated binucleating hydrazone ligand.
4 Experimental
4.1 General remarks
All reactions were accomplished using standard Schlenk-line techniques under an atmosphere of dinitrogen. Solvents were dried and distilled under dinitrogen by standard methods prior to use. Reagents were purchased from commercial sources and used as received. The organometallic hydrazine precursors [CpFe(η6-p-CH3C6H4NHNH2)]+PF6– was synthesized as previously described [27]. The ferrocene-based ketones CpFe(η5-C5H4)CO(p-RC6H4) (R = CH3, H, F) [13], and the diferrocenylketone [28] were prepared according to published procedures. Solid IR spectra were obtained from KBr disks on a Perkin–Elmer, Model Spectrum One, FT IR spectrophotometer. Electronic spectra were recorded in CH2Cl2 and DMSO solutions on a Spectronic, Genesys 2, spectrophotometer. The 1H NMR spectra were recorded on an Avance 400 Digital NMR Bruker spectrometer (400 MHz) at 297 K, and all chemical shifts are quoted in ppm, relative to internal tetramethylsilane (TMS). Cyclic voltammetry experiments were performed at room temperature with a Radiometer PGZ100 potentiostat, using a standard three-electrode setup with a vitreous carbon working and platinum wire auxiliary electrodes and an Ag/AgCl electrode as the reference electrode. Acetonitrile solutions were 1.0 mM in the compound under study and 0.1 M in the supporting electrolyte n-Bu4N+PF6– with a voltage scan rate = 0.1 V s–1. Under these experimental conditions the Cp2Fe/Cp2Fe+ couple was located at 0.507 V (ΔEp = 79 mV). Melting points were determined in evacuated capillaries and were not corrected.
4.2 Preparation of [CpFe(η6-p-CH3C6H4)NHN=C(C6H4-p-CH3)(η5-C5H4)FeCp]+PF6– (2)
A Schlenk tube was charged with a magnetic stirring bar, 50.0 mg (0.129 mmol) of the ionic organometallic hydrazine [CpFe(η6-p-CH3C6H4NHNH2)]+PF6–, 39.2 mg (0.129 mmol) of the toluoylferrocene CpFe(η5-C5H4)–CO(C6H4-p-CH3) (1), and 5.0 ml of ethanol containing 16 μl (0.0129 mmol) of HPF6 0.81 M as the catalyst. The reaction mixture was refluxed for 7 h under dinitrogen. Then, the solvent was evaporated under vacuum, the residue was washed with toluene (3 × 5 ml) to remove the excess of toluoylferrocene, and extracted with methylene chloride (3 × 5 ml). The extracts were combined and concentrated, and addition of an excess of diethyl ether precipitated a solid. Recrystallization from methylene chloride diethyl ether mixture provided 12 mg (0.018 mmol, 14%) of compound 2 as red microcrystals. M.p.: 216 °C. IR (KBr, cm–1): 3289w ν(NH), 3086vw, 2956m, ν(CH), 1559m ν(C=N), 825vs ν(PF6), 554s δ(P–F). 1H NMR (acetone-d6): δ 2.47 (s, 3 H, CH3), 2.54 (s, 3 H, CH3), 4.27 (s, 5 H, CpFe), 4.40 (pseudo-t, 2 H, C5H4), 4.57 (pseudo-t, 2 H, C5H4), 5.03 (s, 5 H, CpFe+), 6.20 (d, 2 H, coord-C6H4, 3JHH = 6.4 Hz), 6.38 (d, 2 H, coord-C6H4, 3JHH = 6.4 Hz), 7.43 (broad s, 4 H, C6H4), 8.71 (s, 1 H, NH). UV–Vis (λmax, nm (log ɛ)): (CH2Cl2) 323 (4.22), 345 sh (4.15), 454 (3.42); (DMSO) 326 (4.14), 343 sh (4.12), 440(3.38).
4.3 X-ray crystal structure determinations
Suitable crystals of compounds 1 and 2 for data collection were selected and mounted with epoxy cement on the tip of a glass fiber. Diffraction intensities were collected with a Bruker SMART APEX diffractometer equipped with a bidimensional CCD detector [29], which employ graphite-monochromated Mo Kα (λ = 0.71073 Å) as radiation source. Crystal, data collection, and refinement parameters are given in Table 4. The two structures were solved using the direct methods, completed by subsequent Fourier syntheses, and refined by full-matrix least-squares procedures on reflection intensities (F2). SADABS absorption corrections were applied to all data. The corresponding space groups were chosen based on the systematic absences in the diffraction data. In the two cases the non-hydrogen atoms were refined with anisotropic displacement coefficients, and all hydrogen atoms were treated as idealized contributions. All software and sources scattering factors are contained in the SHELXTL (5.10) program package [30].
Crystal data collection and structure refinement parameters for compounds 1 and 2
| 1 | 2 | |
| Empirical formula | C18H16FeO | C30H29F6Fe2N2P |
| Formula mass (g mol–1) | 304.16 | 674.22 |
| Collection T (K) | 293(2) | 293(2) |
| Crystal system | Orthorhombic | Triclinic |
| Space group | P212121 | P |
| a (Å) | 9.592(8) | 7.7179(10) |
| b (Å) | 11.177(8) | 10.8954(15) |
| c (Å) | 12.848(9) | 17.131(2) |
| α (°) | 90.0 | 77.810(2) |
| ß (°) | 90.0 | 80.586(2) |
| γ (°) | 90.0 | 86.473(2) |
| V (Å3) | 1377.4(18) | 1388.5(3) |
| Z | 4 | 2 |
| Dcalcd (g cm–3) | 1.467 | 1.613 |
| Crystal size (mm) | 0.55 × 0.25 × 0.15 | 0.80 × 0.50 × 0.50 |
| F(000) | 632 | 688 |
| Absorption coefficient (mm–1) | 1.086 | 1.167 |
| θ Range (°) | 2.65–25.05 | 1.91–28.03 |
| Range h, k, l | 0 ≤ h ≤ 11, 0 ≤ k ≤ 13, 0 ≤ l ≤ 15 | –9 ≤ h ≤ 9, –12 ≤ k ≤ 12, –20 ≤ l ≤ 20 |
| Reflections collected/unique | 1280/1280 | 8459/4799 |
| Data/restraints/parameters | 1280/0/182 | 4799/8/350 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0445, wR2 = 0.1049 | R1 = 0.0848, wR2 = 0.2502 |
| R indices (all data) | R1 = 0.0625, wR2 = 0.1257 | R1 = 0.1448, wR2 = 0.2772 |
| Goodness-of-fit/F2 | 1.104 | 0.938 |
| Largest diffraction peak and hole (e A–3) | 0.532 and –0.868 | 0.480 and –1.333 |
Disorder on the fluorine atoms of PF6 anion in 2 was evident during the last stages of structure completion by difference Fourier synthesis. It was modeled using eight positions whose occupancies were introduced as refinement parameters, and subjected to constrain to add six. Finally, the occupancy of each position (F1: 1.00, F2: 0.60, F3: 0.85, F4: 0.75, F5: 0.75, F6: 0.95, F7: 0.25, F8: 0.85) was held constant during the last stages of refinement. The phosphorous–fluorine (P–F) bond distance was restrained to be 1.58 Å.
5 Supplementary material
Crystallographic data have been sent to the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary publication No. SUP CCDC 207402 for compound 1, and No. SUP CCDC 243680 for compound 2, and can be obtained by contacting the CCDC at http://www.ccdc.cam.ac.uk (quoting the article details and the corresponding SUP number).
Acknowledgements
We thank the financial support from Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT (Chile), Grant No. 1010318 (D.C., C.M.), the CNRS-CONICYT Project No. 16771 (D.C., C.M., J.-R.H.), and the Vicerrectoría de Investigación y Estudios Avanzados, Pontificia Universidad Católica de Valparaíso, Chile.