

1 Introduction
1.1 Background and context
In the field of molecular materials, research into organic light-emitting devices (OLEDs) has been an area of intense and expanding activity over the past decade [1]. Whilst the first generation of such devices employed purely organic polymers, recent work has shown that the incorporation of luminescent d- or f-block metal complexes into these systems can lead to large increases in device efficiency [2]. The high spin–orbit coupling associated with the heavier metal ions promotes emission from triplet excitons formed under the applied electric field, which would otherwise decay through non-radiative processes and severely limit the performance. Complexes of iridium(III) have been at the forefront of recent studies, particularly the tris-cyclometallate fac-[Ir(ppy)3] and its derivatives (ppyH = 2-phenylpyridine), which are both highly emissive and charge-neutral [2b,3]. Photoactive transition metal complexes also offer potential in other areas within the general field of molecular materials, for example, as components of light-activated molecular machines [4], and as photosensitisers of energy and/or electron transfer processes, which may be exploited in the conversion of light to chemical energy [5].
With respect to the incorporation of such complexes into larger supramolecular assemblies, those which contain two terdentate ligands can offer two important advantages over their counterparts with three bidentate ligands. Firstly, the latter are normally chiral, so that a mixture of diastereomers is formed when two or more such complexes are linked together; in contrast, bis-terdentate complexes are achiral. Secondly, complexes with two terdentate ligands have axial symmetry, which allows three or more such components to be connected linearly, and without problems of geometrical isomerism. Such structural advantages of bis-terpyridyl metal complexes over their tris-bipyridyl analogues have been widely recognised [6]. We recently reported (at the Symposium to which this issue is devoted), on the preparation of a charge-neutral, cyclometallated bis-terdentate iridium(III) complex containing N^C^N and C^N^C-coordinating ligands [7]. In parallel with this work, we have been investigating the chemistry of potentially C^N^N-coordinating ligands with iridium(III), and report herein on our preliminary results which reveal that such ligands may bind either as N^N or as N^C-coordinating ligands, according to the structure of the ligand.
1.2 Complexation of 6′-phenyl-2,2′-bipyridine to metal ions
The archetypal ligand for C^N^N-coordination is 6′-phenyl-2,2′-bipyridine, HL1, the complexation of which, by cyclometallation in this manner, was established with Ru(II), Rh(III), Pt(II) and Pd(II) over a decade ago (Fig. 1a) [8]. More recently, Neve and Campagna prepared a series of Ir(III) complexes containing 4′-aryl substituted derivatives of this ligand, of the type shown in Fig. 1b [9]. In the preparation of these complexes, however, the phenylbipyridine ligand is introduced at the final stage, by displacement of chloride from [Ir(ppy)2Cl]2, which rules out the possibility of its binding in a terdentate fashion. In line with the forcing conditions normally required for preparation of tris-cyclometallated complexes of iridium(III), the ligand is found to bind as an N^N rather than as a cyclometallated C^N-coordinating ligand. On the other hand, Mamo and Campagna observed that the ligand 2,6-bis(7′-methyl-4′-phenyl-2′-quinolyl)pyridine (bmpqpy) could bind to iridium either through all three nitrogen atoms (Fig. 2a) or as a C^N^N-coordinating ligand (Fig. 2b), in which ‘roll-over’ cyclometallation of one of the lateral pyridine rings occurs [10].
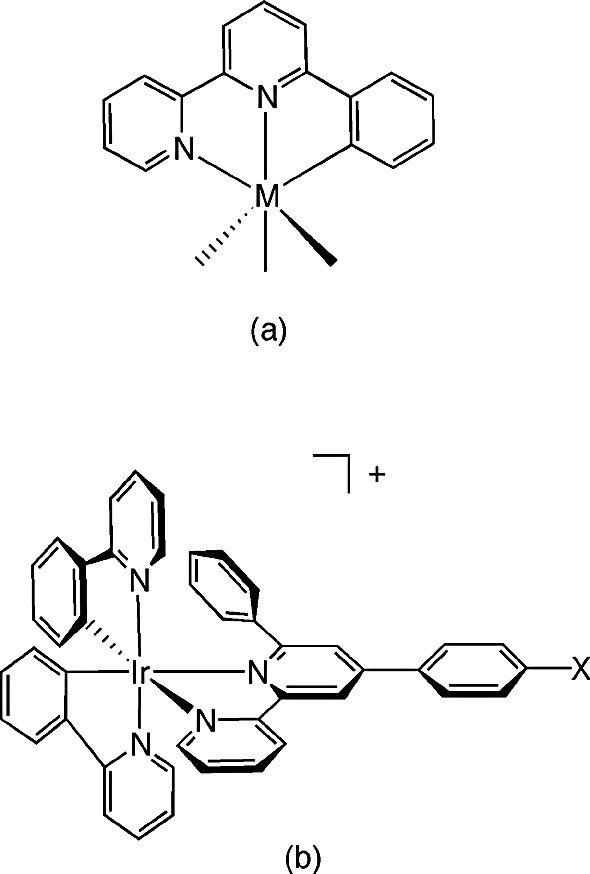
(a) The complexation of 6′-phenyl-2,2′-bipyridine to metal ions as a C^N^N-binding ligand; e.g. M = Ru(II), Pd(II), Pt(II), Rh(III). (b) The N^N-coordination of 6′-phenyl-2,2′-bipyridine derivatives to Ir(III) in previously investigated complexes containing two ppy ligands [9].
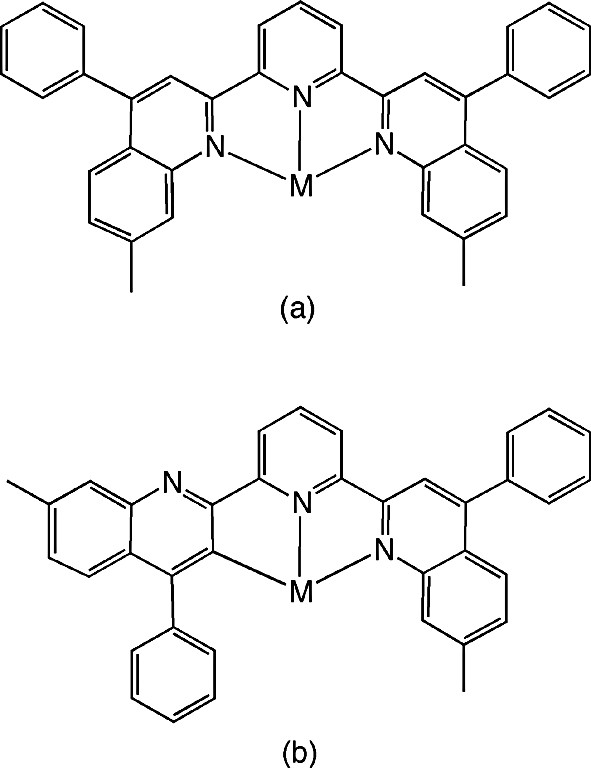
N^N^N versus C^N^N-coordination of 2,6-bis(7′-methyl-4′-phenyl-2′-quinolyl)-pyridine to Ir(III) [10]. Note the presence of the pendent phenyl group ortho to the cyclometalating carbon.
2 Results
2.1 Synthesis of ligands and complexes
We chose to explore the reaction of terpyridyl iridium trichloride, [Ir(tpy)Cl3], with three potentially C^N^N-coordinating ligands. The ligands employed were the parent, unsubstituted ligand (HL1), the 4′-(p-tolyl)-substituted derivative (HL2), and a new, rigidified derivative of the latter, incorporating an ethylene bridge between the central pyridine ring and the lateral phenyl group (HL3) (Fig. 3). Ligand HL1 was prepared by a classical, Kröhnke-type synthesis [11], involving reaction of the enolate equivalent, N-[2-(2-pyridyl)-2-oxoethyl]pyridinium iodide, with the enone equivalent, dimethyl(3-oxo-3-phenylpropyl) ammonium chloride, in the presence of an excess of ammonium acetate in acetic acid [8]. In the case of HL2 and HL3, on the other hand, the requisite 1,5-dicarbonyl compounds were first isolated using the general solvent-free methodology reported by Cave and Raston [12]. Thus, tolualdehyde and acetophenone were ground together in the presence of solid sodium hydroxide to form the α,β-unsaturated ketone, which was then ground with 2-acetylpyridine to give the required 1,5-dicarbonyl compound (Fig. 3). Subsequent reaction with ammonium acetate in ethanol led to HL2 in high yield. This attractive methodology was readily extended to the more rigid system of HL3, by starting from α-tetralone in place of acetophenone (Fig. 3).
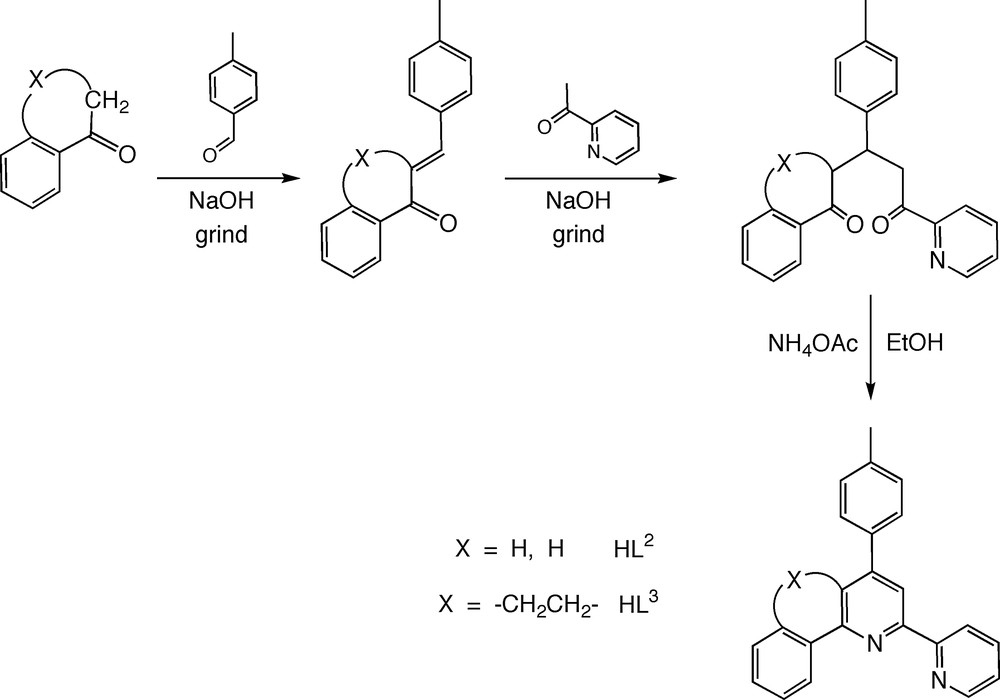
Structures of HL2 and HL3, and the synthetic strategy used to prepare them involving condensation and Michael addition steps under solvent-free conditions.
The reaction of [Ir(tpy)Cl3] with each of the three ligands was carried out in ethylene glycol at reflux. Such forcing conditions have been found to be necessary in the preparation of bis-terpyridyl iridium(III) complexes [13], and generally also for the preparation of fac isomers of tris-cyclometallated iridium compounds [3]. In the case of HL1, the product obtained after 1 h under these conditions, following ion exchange with KPF6 and chromatographic purification, was found to be [Ir(tpy)(HL1)Cl]2+, complex 1 (Fig. 4), in which HL1 binds through its two nitrogen atoms, like bipyridine, but does not cyclometallate to the metal, the pendent phenyl ring remaining unbound. The structure of this complex was elucidated on the basis of 1H NMR spectroscopy (including 1H–1H COSY and NOESY data) and electrospray ionisation mass spectrometry. The twofold symmetry of the pendent phenyl ring of L1 is maintained in the 1H NMR spectrum of the complex, indicating that cyclometallation has not occurred. However, the chemical shifts of the ortho and, to a lesser extent, the meta protons in this ring are low (δ 6.34 and 6.94 ppm, respectively, in CD3CN). This can be accounted for by the fact that the N^N coordination mode will necessarily lead to the positioning of the pendent phenyl ring above the plane of the second, terpyridine ligand, an environment in which the protons of the former will experience an upfield shift, owing to the diamagnetic ring current associated with the latter. Such an effect has been observed previously in ruthenium(II) chemistry, for example, in the structurally related complex [Ru(ttpy)(mapH)Cl]+ (ttpy = 4′-tolylterpyridine, mapH = 2-(p-methoxyphenyl)-1,10-phenanthroline) [14].
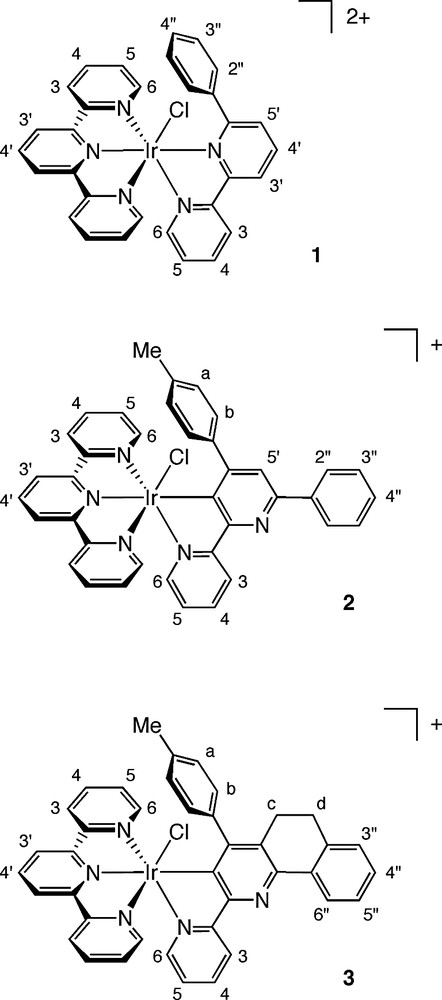
The structures of complexes 1–3.
In an attempt to induce the cyclometallation of the pendent phenyl, the complex was re-heated in ethylene glycol for a longer time (16 h), and in the presence of excess AgBF4. Silver(I) is widely used in Ru(II) polypyridyl chemistry, to favour displacement of coordinated chloride under mild conditions through precipitation of silver chloride. However, in the present instance, the addition of silver(I) had no effect, complex 1 again being recovered. A similar lack of influence of Ag+ on the reaction of [Ir(tpy)Cl3] with terpyridines (to form bis-terpyridyl iridium(III) complexes), has been noted previously [13b], and probably reflects the greater inertness of Ir(III) compared to Ru(II) with respect to ligand substitution reactions. The failure to act as a C^N^N-coordinating ligand, even under forcing conditions, is rather surprising, especially given the relative ease with which HL1 can bind in this way to several other transition metal ions (see above), and the fact that we and others have been able to isolate iridium complexes containing C^N^C-coordinating ligands [7,15a]. Moreover, bis-C^N^N coordination to iridium(III) has been reported very recently, which implies that the terpyridine ligand already present in the coordination sphere of the metal may be inhibiting the cyclometallation of the incoming ligand [15b].
The reaction of [Ir(tpy)Cl3] with ligand HL2, in contrast, led to a cyclometallated product, [Ir(tpy)L2Cl]+. However, rather than the anticipated C^N^N-coordination, analysis of the one- and two-dimensional 1H NMR spectra revealed the product to be complex 2, in which the central pyridine ring of L2 has cyclometallated through C3′, rather than binding to the metal ion through its nitrogen atom. Twofold symmetry in the pendent phenyl and tolyl rings is maintained in the 1H NMR spectrum of the complex, confirming that neither of these aryl rings has undergone cyclometallation. In this case, however, it is the pendent tolyl ring, rather than the phenyl ring, that shows the upfield shifts that are characteristic of a ring being positioned above the plane of the Ir(tpy) fragment: the phenyl ring resonances appear at higher frequencies. This is indicative of the N^C-binding mode shown (Fig. 4), which is further supported by the 1H–1H NOESY data.
The rigidified ligand HL3 reacted with [Ir(tpy)Cl3] under comparable conditions to give a similar product, [Ir(tpy)L3Cl]+, in which the central pyridine ring is again cyclometallated, leading to the positioning of the tolyl moiety above the Ir(tpy) plane, as above. It is interesting to note that the annelated ring system in HL3 apparently does nothing to favour the cyclometallation of the phenyl group which would lead to the C^N^N configuration.
The differing behaviour of ligands HL2 and HL3 compared to HL1, namely N^C coordination of the former rather than N^N in the latter, is intriguing. Since the only difference between HL1 and HL2 is the presence of the 4′-tolyl substituent in HL2, it suggests that this aryl substituent may activate the adjacent position of the pyridyl ring (C3′) to cyclometallation, whereas, in its absence, coordination through nitrogen is more favourable. It is noteworthy that in the C^N^N-coordinated iridium(III) complexes of the bmpqpy ligand discussed earlier, where “roll-over” cyclometallation of a lateral pyridyl ring was observed (Fig. 2b), the site of cyclometallation also has an aryl group in the position adjacent to it [10]. Possibly, the formal deprotonation of the carbon atom, which accompanies cyclometallation, is promoted through conjugation with the ortho-disposed phenyl substituent.
2.2 Photophysical properties of the complexes
The absorption spectra of the complexes in acetonitrile solution at room temperature are shown in Fig. 5 (see also Table 1). The spectra of complexes 2 and 3 are very similar to one another, consistent with their similar structures and the N^C-binding mode adopted in both cases. On the other hand, their spectra are quite different from that of complex 1. In particular, the former display strong bands around 380 nm, together with weaker, but distinct bands in the visible, between 450 and 500 nm, which are absent in the spectrum of 1. These bands are most likely due to absorption to 1MLCT and 3MLCT states, respectively. It is well-established in the chemistry of iridium(III) with polyimine ligands that the presence of anionic co-ligands, such as chloride or a cyclometallating carbon, serves to lower the energy of MLCT states, by compensating for metal-centred oxidation [16]. In contrast, in complexes with exclusively neutral ligands, there is insufficient donation of electron density to the metal to favour MLCT, such that the lowest energy excited states in complexes like [Ir(bpy)3]3+ are LC in character, with the MLCT states lying much higher in energy [13,16]. In the present instance, the presence of both a chloride and a cyclometallating carbon in the coordination sphere of 2 and 3 should stabilise the MLCT process, leading to the observed low-energy bands. Indeed, the absorption spectra of these two complexes are similar to the spectra normally displayed by iridium complexes with a (C^N)2(N^N) coordination sphere (i.e. complexes which also contain two anionic ligating atoms), such as [Ir(ppy)2(bpy)]+ [17]. The electron density on the metal ion in complex 1, on the other hand, is expected to be lower (as there is no cyclometallating carbon present), and metal-centred oxidation correspondingly less favourable, leading to the absence of low-energy, charge-transfer bands.
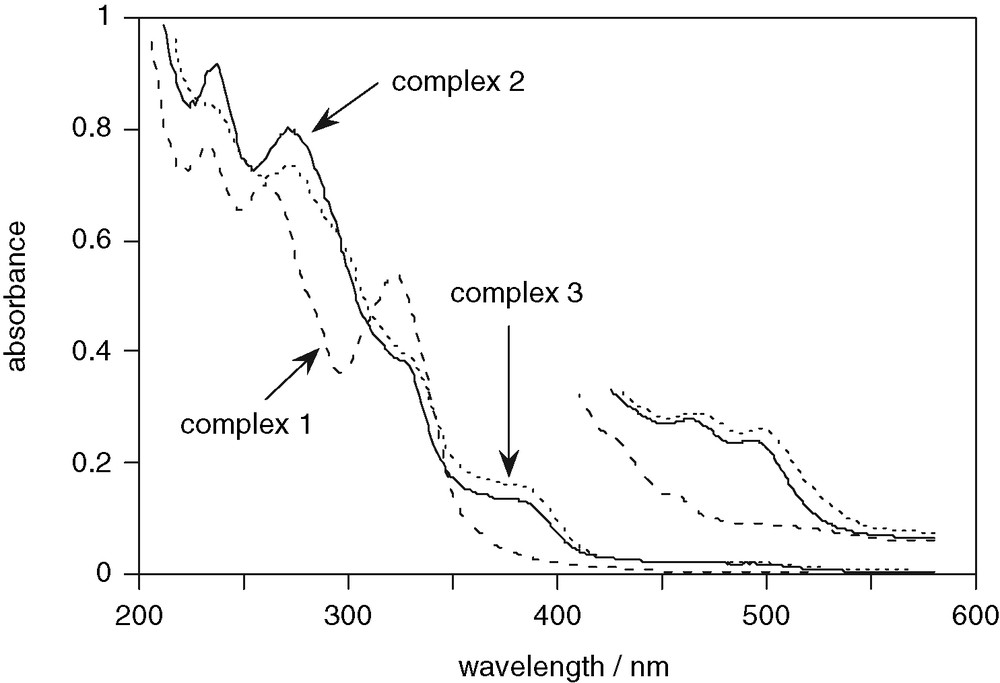
UV–visible absorption spectra of [Ir(tpy)HL1Cl]2+ (1, dashed line), [Ir(tpy)L2Cl]+ (2, solid line), and [Ir(tpy)L3Cl]+ (3, dotted line), in acetonitrile solution at 295 K. The weak, long wavelength bands are shown on an expanded scale for clarity.
Photophysical parameters of the iridium(III) complexesa
| Complex 1 [Ir(tpy)HL1Cl]2+ | Complex 2 [Ir(tpy)L2Cl]+ | Complex 3 [Ir(tpy)L3Cl]+ | |
| 237 (50 100) | 234 (31 300) | ||
| Absorption | 233 (36 400) | 271 (43 700) | 272 (25 600) |
| λmax (nm) (ɛ) | 261 (33 200) | 328 sh (19 900) | 330 sh (14 100) |
| 322 (25 800) | 376 (6 500) | 381 (5 600) | |
| 463 (900) | 467 (900) | ||
| 494 (700) | 496 (700) | ||
| Emissionb λmax (nm) | 539 | 541 | 562 |
| τc (μs) degassed (aerated) | 1.5 (0.8) | 3.2 (1.5) | 2.4 (0.8) |
| 102 × φd degassed (aerated) | 0.70 (0.31) | 16 (7.9) | 16 (5.4) |
| kQe × 10–8 (mol–1 dm3 s–1) | 3.1 | 1.8 | 4.4 |
a In CH3CN at 295 K.
b λex = 360, 376, 381 nm for 1–3, respectively.
c λex = 355 nm; estimated uncertainty ± 10%.
d Measured using an excitation wavelength of 355 nm and band-passes of 2.5 nm; quinine sulphate was used as the standard (ϕ = 0.546 in 1 M H2SO4 [19]); estimated uncertainty ± 15%.
e Estimated on the basis of lifetime data in deoxygenated solution and at atmospheric pressure of air. At a partial pressure of 0.21 atm, [O2] = 1.9 mmol dm–3 in CH3CN [20].
All three complexes are luminescent in acetonitrile solution at room temperature, with luminescence lifetimes in the microsecond range (Fig. 6 and Table 1). Complexes 2 and 3 are strongly emissive, with quantum yields of 16% in degassed solution. Their spectra exhibit similar profiles, although the latter displays a significant red-shift of 20 nm compared to the former. The broad, structureless nature of the emission, together with the microsecond lifetimes, is consistent with an emissive excited state of primarily charge-transfer character, as generally observed also for complexes with a (C^N)2(N^N) coordination sphere [17,18]. The red-shift in the emission of complex 3 is then to be anticipated, since the ethylene bridge in the annelated ligand will favour a roughly planar conformation of the pendent phenyl group with respect to the central pyridine ring, thereby assisting conjugation between them, lowering the energy of the acceptor π* orbitals, and hence the emission energy. The redshift is accompanied by a shortening of the excited state lifetime from 3.2 μs for 2 to 2.4 μs for 3 (degassed acetonitrile solution). The luminescence of both complexes is quenched to a modest extent by dissolved molecular oxygen, the bimolecular rate constant of quenching being rather higher for 3 than for 2, possibly owing to the more rigid nature of the former.
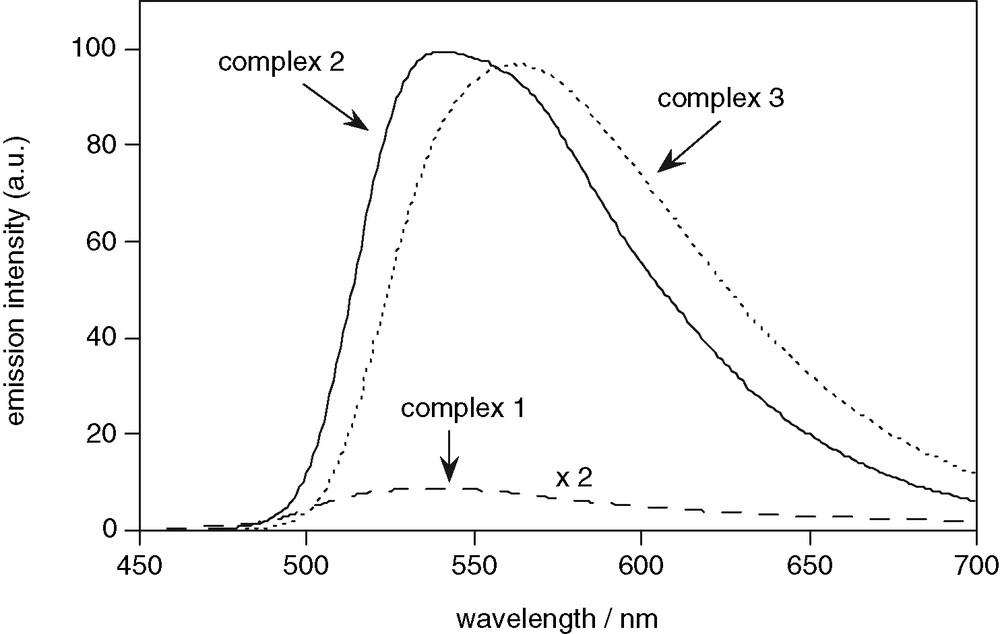
Luminescence emission spectra of [Ir(tpy)HL1Cl]2+ (1, dashed line), [Ir(tpy)L2Cl]+ (2, solid line), and [Ir(tpy)L3Cl]+ (3, dotted line), for isoabsorbant solutions in acetonitrile solution at 295 K; λex = 380 nm, band-passes = 2.5 nm.
Complex 1, on the other hand, is only weakly emissive under the same conditions, with a quantum yield over 20 times smaller than the values for the cyclometallated complexes (Fig. 6 and Table 1). A pertinent comparison to make in this case is with [Ir(tpy)2]3+, which displays a highly structured 3LC emission, λmax = 491 nm in CH3CN [13b]. The red-shifted luminescence of 1 (λmax = 539 nm) relative to the bis-terpyridyl analogue suggests that the 6′-phenyl-bipyridine ligand makes a significant contribution to the HOMO and/or LUMO in 1, and possibly also that the presence of the chloride ligand confers more charge-transfer character to the excited state than in [Ir(tpy)2]3+.
3 Conclusions
The development of transition metal complexes which display intense luminescence from triplet-excited states is currently an important theme within molecular materials research, as indicated in the introduction. In the case of iridium(III), cyclometallated complexes are important, yet few such compounds have been reported that contain terdentate ligands, despite the structural advantages offered over the widely investigated tris-bidentate systems. The present work reveals that simple 6′-phenyl-2,2′-bipyridines show no propensity to bind to Ir(III) as C^N^N-coordinating ligands, in the presence of terpyridine as the second, N^N^N-coordinated ligand. On the other hand, unexpected cyclometallation via the central pyridine ring can occur, to give N^C coordination, apparently activated by ortho-disposed aryl substituents. The resulting monocationic complexes are strongly luminescent in solution at room temperature, auguring well for further investigations into the chemistry and photophysics of such compounds.
4 Experimental
4.1 Synthetic details
Solvents used were Analar quality or HPLC-grade; water was purified using the Purite Still-plus system. 1H NMR spectra were recorded on a Varian 400 or 500 MHz instrument, and referenced to residual protio-solvent resonances. Coupling constants are in Hertz. The numbering system for NMR assignments is given in Fig. 4. FAB ionisation mass spectra were recorded at the EPSRC National Mass Spectrometry Service Centre, Swansea. HL1 was prepared by Kröhnke [11] methodology, using a procedure similar to that described previously [8]. Terpyridyl iridium trichloride was prepared from terpyridine and iridium trichloride trihydrate, as reported previously [13b].
4.1.1 4′-(p-Tolyl)-6′-phenyl-2,2′-bipyridine HL2
The synthesis of this compound was based on the general methodology reported recently by Raston, namely a variant of the Kröhnke procedure, in which the initial condensation steps are carried out in the absence of solvent [12]. Acetophenone (3.00 g, 25 mmol), p-tolualdehyde (3.00 g, 25 mmol) and NaOH pellets (0.99 g, 24.8 mmol) were ground together using a pestle and mortar for 15 min, until a pale yellow powder of 1-phenyl-3-(4′-tolyl)-propen-1-one was formed. A portion of this compound (3.79 g, 17.1 mmol) was then ground with 2-acetylpyridine (2.07 g, 17.1 mmol) and more NaOH pellets (0.68 g, 17.0 mmol) for 10 min to afford the dicarbonyl compound, 1-phenyl-5-(pyridin-2-yl)-3-(p-tolyl)-pentan-1,5-dione, as a pale yellow solid. The crude product was dissolved in ethanol (60 ml), ammonium acetate added (5.0 g, 65 mmol), and the mixture heated to reflux for 3 h. Pale yellow crystals formed upon cooling, which were separated and washed with cold ethanol, followed by diethyl ether. Further product could be obtained by concentrating the mother liquor further. The total isolated yield was 0.66 g (12% overall). NMR data is consistent with that for the compound prepared by a more conventional route [9a]; the present data was obtained at higher frequency, allowing a full assignment to be made. 1H NMR (500 MHz, CDCl3): δ 8.73 (1H, dd, J 4.7, 0.7, H6), 8.69 (1H, d, J 8.0, H3), 8.64 (1H, d, J 1.5, H3′), 8.21 (2H, d, J 7.5, H2″), 7.98 (1H, d, J 1.5, H5′), 7.87 (1H, td, J 7.7, 1.8, H4), 7.75 (2H, d, J 8.0, Hb), 7.53 (2H, t, J 7.5, H3″), 7.46 (1H, tt, J 7.0, 2.0, H4″), 7.35 (1H, td, J 4.5, 1.7, H5), 7.33 (2H, d, J 8.0, Ha), 2.44 (3H, d, Me). MS (EI): m/z 322 (M+), 307 (M+–Me), 244 (M+–py). Anal. calcd for C23H18N2: C, 85.68, H, 5.63, N, 8.69%. Found: C, 85.13, H, 5.79, N, 8.44%.
4.1.2 2-(2-Pyridyl)-4-(p-tolyl)-5,6-dihydro-7,8-benzoquinoline HL3
This compound was prepared using a similar method to that described above for HL2. α-Tetralone (3.00 g, 21.4 mmol), p-tolualdehyde (2.57 g, 21.4 mmol) and NaOH pellets (0.86 g, 21.5 mmol) were ground together with a pestle and mortar for 15 min to give the α,β-unsaturated ketone, 2-(1-p-tolyl-methylidene)-3,4-dihydronaphthalen-1-one, as an off-white powder. 1H NMR (300 MHz, CDCl3): δ 8.13 (1H, dd, J 7.5, 0.9, H8), 7.86 (1H, s, Ha), 7.49 (1H, td, J 8.2, 1.5, H6), 7.41–7.32 (3H, m, H2′ and H7), 7.30–7.20 (3H, m, H3′ and H5), 3.14 (2H, td, J 6.9, 1.5, CH2d), 2.94 (2H, t, J 6.9, CH2c), 2.39 (s, 3H, Me). This compound was then ground with 2-acetylpyridine (2.59 g, 21.4 mmol) and more NaOH (0.86 g, 21.5 mmol) for 25 min, to give the 1,5-dicarbonyl compound (Fig. 3). A mixture of the crude product and ammonium acetate (6.0 g, 78 mmol) in ethanol (60 ml) was heated at reflux for 19 h. Upon cooling, the precipitated solid was separated and washed with cold ethanol followed by diethyl ether, to give 2.76 g product (37%). A sample of material was recrystallised from ethanol; m.p. 140 °C. 1H NMR (500 MHz, CDCl3): δ 8.68 (1H, d, J 8.0, H3), 8.66 (1H, d, J 7.5, H6), 8.56 (1H, d, J 10.0, H6″), 8.29 (1H, s, H3′), 7.85 (1H, td, J 8.0, 1.5, H4), 7.42 (1H, t, J 7.5, H5″), 7.39–7.31 (3H, m, H4″ and Ha), 7.31–7.26 (2H, m, H5 and Hb), 7.26–7.22 (1H, m, H3″), 2.99 (2H, t, J 6.5, CH2c), 2.87 (2H, t, J 6.5, CH2d), 2.43 (3H, s, Me). MS (EI): m/z 348 (M+), 347 (M+ – H). Anal. calcd for C25H20N2: C, 86.16, H, 5.79, N, 8.05%. Found: C, 85.55, H, 5.98, N, 7.51%.
4.1.3 [Ir(tpy)HL1Cl](PF6)2 – complex 1
A mixture of [Ir(tpy)Cl3] (229 mg, 0.43 mmol) and HL1 (100 mg, 0.43 mmol) in ethylene glycol (5 ml) was heated at reflux under a nitrogen atmosphere for 1 h to give a clear red solution. The solution was added dropwise to a saturated aqueous solution of KPF6, and the resulting precipitate was separated and washed with water to give the crude product as a yellow solid (301 mg, 71%). A sample of the compound was purified by column chromatography on silica, gradient elution from CH3CN to 80% CH3CN/19% H2O/1% KNO3 (aq), Rf (TLC) = 0.4 in the latter solvent system, followed by ion exchange with KPF6 to give complex 1 as a yellow solid. 1H NMR (500 MHz, MeCN): δ 9.76 (1H, d, J 6.0, H3), 8.83 (1H, d, J 8.5, H6), 8.62 (1H, dd, J 8.0, 1.0, H3′), 8.59 (1H, td, J 8.0, 1.5, H5), 8.35 (2H, d, J 7.5, H3tpy), 8.23 (2H, td, J 8.0, 1.5, H4tpy), 8.19 (2H, d, J 8.0, H3′tpy), 8.11–8.05 (3H, m, H4 + H4′ + H4′tpy), 7.75 (2H, dd, J 5.5, 1.0, H6tpy), 7.59 (2H, td, J 7.0, 1.5, H5tpy), 7.27 (1H, t, J 7.5, H4″), 7.16 (1H, dd, J 7.5, 1.5, H5′), 6.94 (2H, t, J 8.0, H3″), 6.34 (2H, dd, J 8.5, 1.5, H2″). MS (ES+): m/z 346.7 (M2+), 838.3 (M + PF6)+. HRMS (ES+): 346.5609 (M2+); calcd for C31H23N535Cl193Ir, M2+: 346.5638.
4.1.4 [Ir(tpy)L2Cl](PF6) – complex 2
A mixture of [Ir(tpy)Cl3] (195 mg, 0.37 mmol) and HL2 (118 mg, 0.37 mmol) in ethylene glycol (5 ml) was heated at reflux under a nitrogen atmosphere for 16 h. Upon cooling, the solution was diluted with water (13 ml), and a small amount of an insoluble material removed. The resulting dark red solution was added to saturated aqueous KPF6, and the precipitate formed was separated and washed with water to give the crude product (190 mg, 55%). Purification of a sample of the material was achieved by chromatography on silica, gradient elution from CH3CN to 88% CH3CN/11% H2O/1% KNO3 (aq), Rf (TLC) = 0.5 in the latter solvent system, followed by further ion exchange of appropriate fractions with KPF6, to give complex 2 as a yellow–orange solid. 1H NMR (500 MHz, d6-acetone): δ 10.11 (1H, d, J 5.0, H3), 8.95 (1H, d, J 8.5, H6), 8.60 (2H, d, J 8.0, H3tpy), 8.46 (1H, td, J 8.0, 1.0, H5), 8.41 (2H, d, J 8.5, H3′tpy), 8.28 (2H, td, J 7.5, 1.5, H4tpy), 8.26 (1H, t, J 8.0, H4′tpy), 8.17 (2H, d, J 7.0, H2″), 7.91 (2H, d, J 5.5, H6tpy), 7.89 (1H, m, H4), 7.63 (2H, td, J 7.0, 1.0, H5tpy), 7.46 (2H, t, J 7.5, H3″), 7.40 (1H, t, J 7.5, H4″), 7.05 (1H, s, H5′), 6.77 (2H, d, J 7.5, Ha), 6.17 (2H, d, J 7.5, Hb), 2.33 (3H, s, Me). MS (ES+): m/z 782.2 (M+), 373.6 (M – Cl)2+. HRMS (ES+): 782.1680 (M+); calcd for C38H28N535Cl193Ir, M+: 782.1667.
4.1.5 [Ir(tpy)L3Cl](PF6) – complex 3
A procedure similar to that described above for complex 2 was employed. A mixture of [Ir(tpy)Cl3] (153 mg, 0.29 mmol) and HL3 (100 mg, 0.29 mmol) in ethylene glycol (5 ml) was heated at reflux under a nitrogen atmosphere for 17 h. Upon cooling, the solution was diluted with water and filtered through a bed of celite to remove a fine dark precipitate. The orange–yellow solution was then added dropwise to an aqueous solution of KPF6, and the yellow precipitate formed was separated and washed with water to give the crude product (103 mg, 37%). Purification of a sample of the material was achieved by chromatography on silica, gradient elution from CH3CN to 88% CH3CN/11% H2O/1% KNO3 (aq), Rf (TLC) = 0.5 in the latter solvent system, followed by further ion exchange of appropriate fractions with KPF6, to give complex 3 as a yellow solid. 1H NMR (500 MHz, d6-acetone): δ 9.94 (1H, d, J 6.0, H3), 8.80 (1H, d, J 7.5, H6), 8.45 (2H, d, J 7.5, H3tp), 8.39 (1H, d, J 7.5, H6″), 8.30 (1H, t, J 8.0, 1.5, H5), 8.27 (2H, d, J 8.0, H3′tpy), 8.17–8.11 (3H, m, H4tpy + H4′tpy), 7.77 (2H, d, J 5.0, H6tpy), 7.71 (1H, td, J 6.5, 1.5, H4), 7.50 (2H, td, J 6.7, 1.5, H5tp), 7.24 (1H, t, J 7.5, H5″), 7.15 (1H, td, J 7.5, 1.5, H4″), 7.00 (1H, d, J 7.5, H3″), 6.65 (2H, d, J 7.5, Ha), 5.94 (2H, d, J 7.5, Hb), 2.43 (2H, m, CH2d), 1.92 (2H, m, CH2c). MS (ES+): m/z 808.3 (M+), 386.8 (M – Cl)2+. HRMS (ES+): 808.1989 (M+); calcd for C40H30N535Cl193Ir, M+: 808.1973.
4.2 Photophysical measurements
Absorption spectra were measured using a Biotek Instruments XS spectrometer, using matched quartz cuvettes of 1 cm pathlength. Luminescence excitation and emission spectra were recorded using a Jobin Yvon FluoroMax-2 spectrofluorimeter, equipped with a Hamamatsu R928 photomultiplier tube. Samples for time-resolved measurements were excited at 355 nm using the third harmonic of a Q-switched Nd:YAG laser, and the luminescence detected with a Hamamatsu R928 photomultiplier tube and recorded using a digital storage oscilloscope, before transfer to a PC for analysis; estimated uncertainty in lifetimes is ± 10% or better. Samples were degassed via a minimum of three freeze–pump–thaw cycles, base pressure < 10–3 mbar, in 1-cm pathlength quartz cuvettes modified to allow connection to the vacuum line.
Acknowledgements
We thank the University of Durham for support, Dr A. Beeby for use of the Nd:YAG laser, and the EPSRC National Mass Spectrometry Service Centre, Swansea.
Vous devez vous connecter pour continuer.
S'authentifier