

1 Introduction
Previous experimental surface studies of the adsorption of perfluorohexane and perfluoroheptane on single-crystal Pt(111) surfaces under ultra-high vacuum conditions [1] showed that the thermal desorption spectra (TDS) of both perfluoroalkanes were similar. That is, each perfluoroalkane exhibited two distinct desorption peaks, denoted α1 and α2 in that study, due to monolayer species. At low surface coverages, the higher binding energy surface species, α1, was believed to lie ‘flat’ on the Pt surface, thereby maximizing adsorbate–surface interactions for the molecule. The unexpected lower binding energy surface species, α2, had a binding energy thought to be consistent with a tilted or puckered configuration in which the interaction of the molecule with the surface was reduced. The decrease in surface interaction, and thus binding energy, for the α2 species was thought to be consistent with the binding of at least one CF2 unit less than that of the α1 species.
While the infrared reflection–absorption spectra (IR-RAS or IRAS) of these monolayers indicated an orientation of the perfluoroalkanes on the metal surface that was different from that of the bulk condensate layer, nothing could be conclusively determined regarding the relative orientations of the α1 and α2 species relative to the metal surface. The IR-RAS results could not be unambiguously interpreted due to mixing of the C–F functional group frequencies with the C–C skeletal modes. Because of this complication, structures of the α1 and α2 species could not be assigned unambiguously.
In the experimental study [1], it was assumed that the lowest-energy conformer of the perfluoroalkane (of unknown structure) was the conformer that adsorbed on the metal surface as the α1 species, and that the structures of these lowest-energy perfluoroalkane conformers were analogous to the structures of the corresponding alkanes. This meant that the perfluorocarbon species adsorbed on a Pt(111) surface would be derived from the lowest-energy, staggered, zig–zag alkane conformer. However, poly(tetrafluoroethylene), PTFE, is well-known to adopt a helical structure [2,3], and, despite the challenges of carrying out high-level quantum mechanical calculations on perfluorinated hydrocarbons, significant computational results [4–7] indicate that smaller perfluorocarbons also adopt helical structures or structures that would be helical if the molecular chains were extended [6]. (A recent, comprehensive literature search indicated that no direct experimental investigations of the adsorption geometries of perfluorocarbon molecules on surfaces, using LEED for example, have been reported).
A limited number of groups have been extensively involved in computational perfluoroalkane conformational analyses [4–8]. Not only does poly(tetrafluoroethylene) adopt a helical structure, but a fully helical structure has been shown to be the lowest-energy conformer for perfluoropentane, -hexane, and -heptane at the Hartree–Fock RHF/3-21G(*) level of theory [9]. This result is confirmed by the novel force-field and density functional calculations recently reported by Jang et al. [10] for a variety of perfluoroalkanes.
The comprehensive conformational analyses of perfluoropentane, -hexane, and -heptane [9], in confirming the lowest-energy helical structures of these molecules, also resulted in elucidation of the conformational manifold exhibited by these three molecules. Significantly, both the lowest-energy totally helical conformer and the next-lowest-energy semi-helical (“kinked”) conformers of perfluorohexane and perfluoroheptane have important implications for the interpretation of the thermal desorption behavior of these molecules on a Pt(111) surface. With the goal of explaining the high-coverage dual-state adsorption behavior of perfluoroalkanes on Pt(111) surfaces, the two lowest-energy conformers of perfluorohexane and perfluoroheptane were re-investigated at the Hartree–Fock RHF/6-31G* level of theory. In addition, with the α1 and α2 surface species in mind, a transition state for the interconversion of these lowest-energy conformers was identified and investigated at this same level of theory.
Herein are reported the computational results for the two lowest-energy conformers of perfluorohexane and perfluoroheptane, and a transition state defining the interconversion of the conformers for each molecule. These results are then applied to the interpretation of the previously-reported thermal desorption data to aid in clarification and refinement of previously proposed surface structures [1] for perfluorohexane and perfluoroheptane on a Pt(111) surface.
2 Computational methods
The PC Spartan Pro® computational package [11] was used for the computational determination of all ground and transition state molecular properties. The two lowest-energy conformers of perfluorohexane and perfluoroheptane were identified from detailed RHF/3-21G(*) conformational manifolds of the molecules [9], and these conformers were optimized at the RHF/6-31G* level of theory (including complete vibrational analyses to confirm that the molecules were true ground state minima). As the two lowest-energy conformers for each molecule differ only by rotation of approximately 120° about one C–C bond, a potential transition state structure for the interconversion between the conformers (and perhaps α1 and α2 surface structures) was the eclipsed conformer along this rotational pathway. Transition state optimizations of these structures, followed by complete vibrational analyses, were carried out at the RHF/6-31G* level of theory as well.
3 Results and discussion
The structures of the lowest-energy conformers of perfluorohexane and perfluoroheptane are illustrated in Figs. 1 and 2, respectively, as both ball-and-stick and space-filling models. As these illustrations demonstrate, the helicity of the lowest-energy conformers (Ahex and Ahept) is evident in the space-filling models, while the ‘kinked’ nature of the next lowest-energy conformers (Bhex and Bhept) is equally apparent. A pitch angle of approximately 20° is observed in each helical molecule (Ahex and Ahept). The higher energy B conformers are related to the lower energy A conformers by a rotation of approximately 120° about the C3–C4 bond of perfluorohexane or about the C4–C5 bond of perfluoroheptane, as illustrated by the Newman projections in Fig. 3.
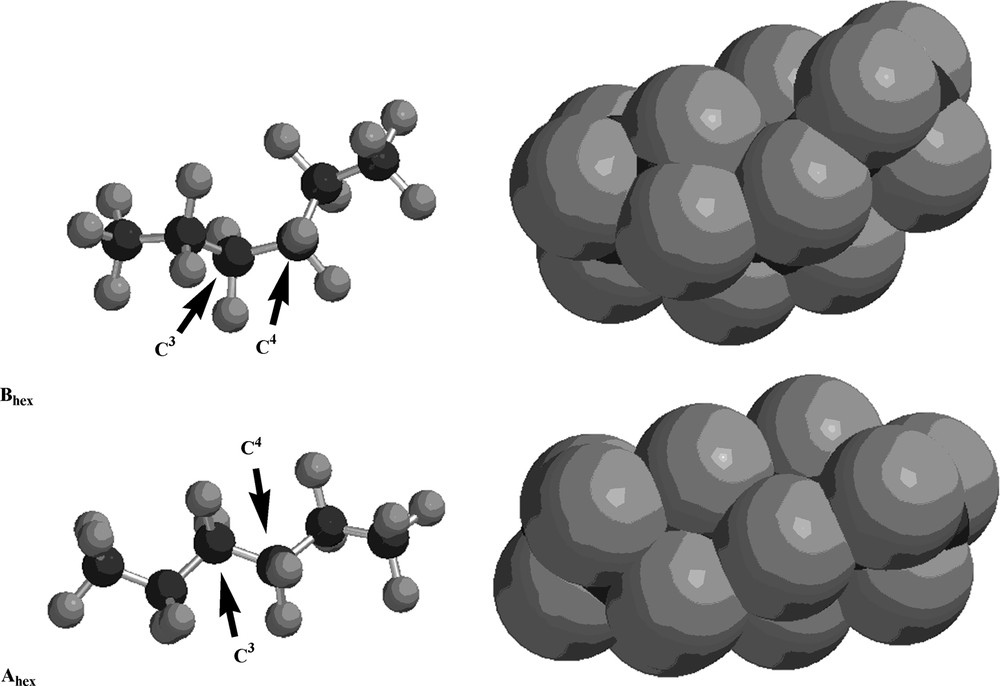
The structures of the lowest-energy (Ahex) and next lowest-energy (Bhex) conformers of perfluorohexane as both ball-and-stick and space-filling models. Black spheres are carbon atoms; gray spheres are fluorine atoms. The arrows indicate C3 and C4, respectively.
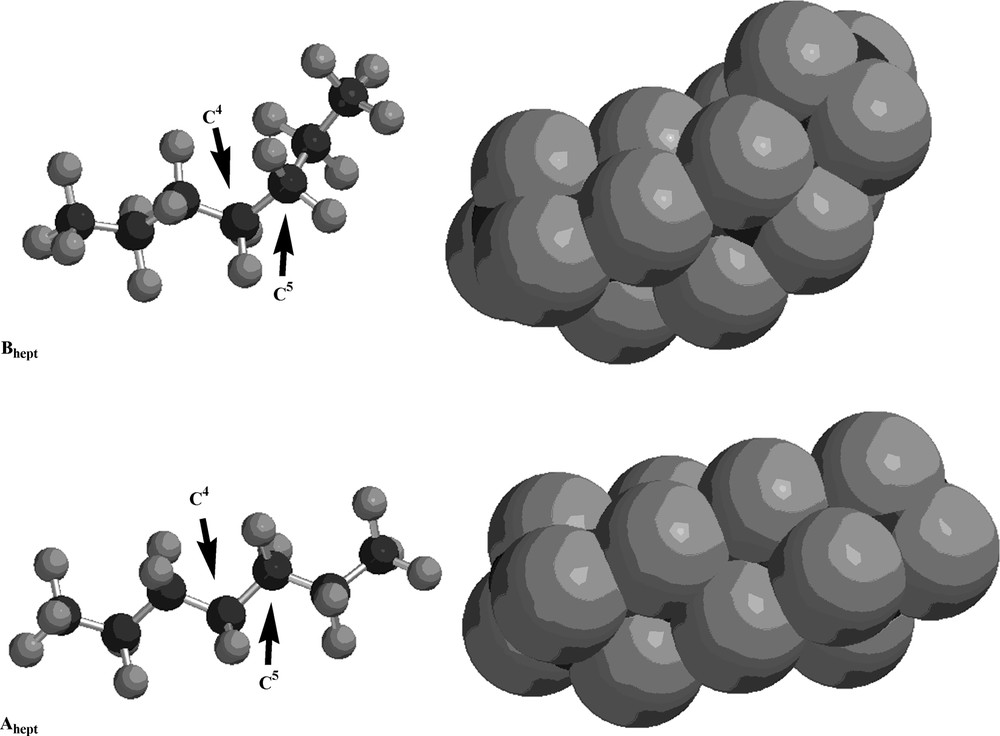
The structures of the lowest-energy (Ahept) and next lowest-energy (Bhept) conformers of perfluoroheptane as both ball-and-stick and space-filling models. Black spheres are carbon atoms; gray spheres are fluorine atoms. The arrows indicate C4 and C5, respectively.
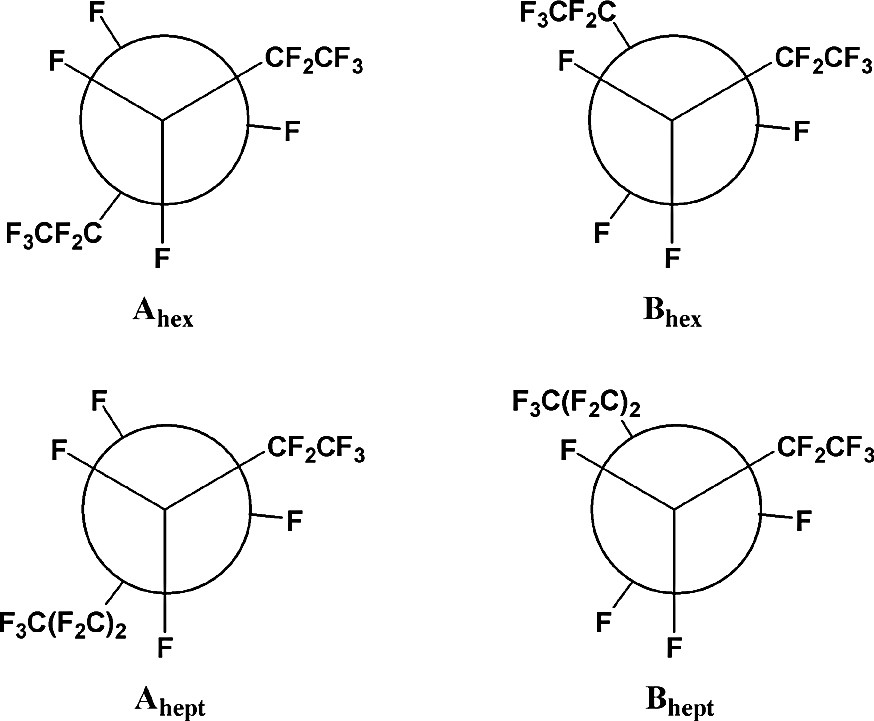
Newman projections of the two lowest-energy conformers of perfluorohexane and perfluoroheptane, showing that the conformers differ only by a rotation of approximately 120° about the C3–C4 bond of perfluorohexane or the C4–C5 bond of perfluoroheptane.
The calculated properties for the two lowest-energy conformers (conformer A is lower in energy than conformer B) of perfluorohexane and perfluoroheptane, as well as the calculated properties of the proposed transition states for the interconversion between the two lowest-energy conformers, are listed in Table 1. It is noteworthy that the difference in energy between the two lowest-energy conformers for each perfluoroalkane is approximately the same (~8.6 kJ mol–1). The energy difference between each of the ground state conformers and the transition state for the interconversion between them is also of a similar magnitude, but the energy differences between the ground state conformers and the transition state for perfluoroheptane are approximately 2 kJ mol–1 greater than the analogous differences for perfluorohexane.
Calculated properties for perfluorohexane and perfluoroheptane conformers and transition states
| Moleculea | Hartree–Fock energyb | HF energy difference | HOMO energyc | LUMO energyd | Dipole momente |
| Perfluorohexane | |||||
| Conformer Ahex | –1619.3473341 a.u. | –14.301 eV | +4.997 eV | 0.072 D | |
| Conformer Bhex | –1619.3440585 a.u. | –14.472 eV | +5.194 eV | 0.029 D | |
| EB – EA | 8.600 kJ mol–1 | ||||
| TS Ahex ↔ Bhex | –1619.3422884 a.u. | –14.374 eV | +5.018 eV | 0.091 D | |
| ETS – EA | 13.247 kJ mol–1 | ||||
| ETS – EB | 4.647 kJ mol–1 | ||||
| Perfluoroheptane | |||||
| Conformer Ahept | –1856.0879490 a.u. | –14.158 eV | +4.716 eV | 0.091 D | |
| Conformer Bhept | –1856.0846497 a.u. | –14.310 eV | +4.898 eV | 0.138 D | |
| EB – EA | 8.662 kJ mol–1 | ||||
| TS Ahept ↔ Bhept | –1856.0821294 a.u. | –14.200 eV | +4.670 eV | 0.147 D | |
| ETS – EA | 15.279 kJ mol–1 | ||||
| ETS – EB | 6.617 kJ mol–1 |
a The abbreviation TS refers to the transition state structure; the Hartree–Fock energy differences between the conformers are symbolized EB – EA, while the energy differences between conformers A or B and the transition state structure are denoted using the symbols ETS – EA and ETS – EB, respectively.
b The symbol a.u. refers to an atomic unit; 1 a.u. = 627.510 kcal mol–1 = 1625.50 kJ mol–1.
c HOMO is the abbreviation for highest occupied molecular orbital; the energy is given in electron volts, eV, where 1 eV = 96.4853 kJ mol–1.
d LUMO is the abbreviation for the lowest unoccupied molecular orbital.
e Dipole moments are given in units of the debye, D; 1 D = 3.33564 × 10–30 C m.
The transition state structure for the interconversion between the perfluorohexane conformers (TShex) is depicted in Fig. 4, and Fig. 5 illustrates the transition state structure for the perfluoroheptane conformer interconversion (TShept). As expected, the transition state structure for the interconversion of the two lowest-energy conformers of each perfluorocarbon is an eclipsed structure, as depicted by the Newman projections in Figs. 4 and 5.
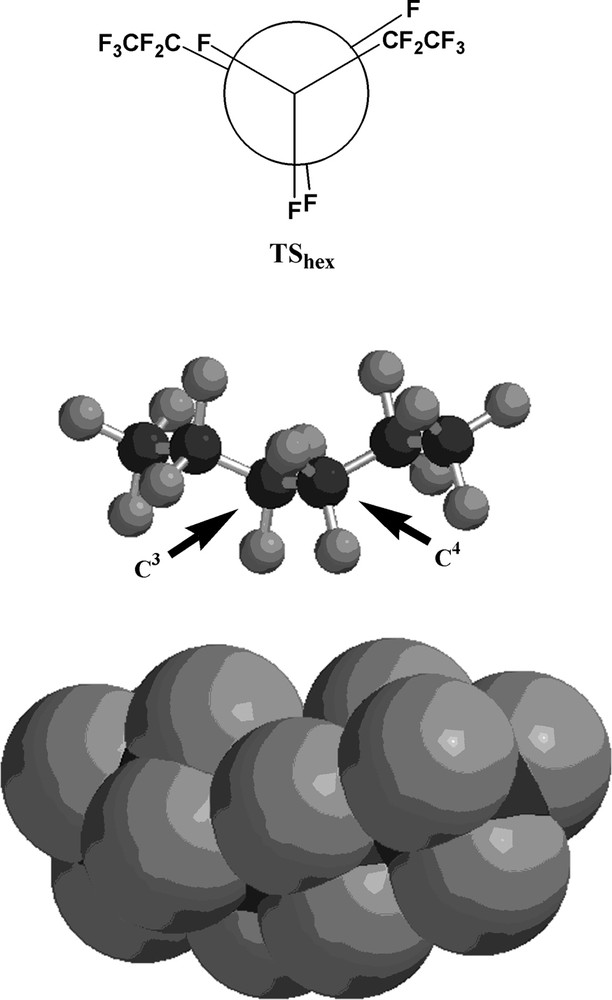
The transition state structure (TShex) for the interconversion of the two lowest-energy conformers (Ahex and Bhex) of perfluorohexane. Black spheres are carbon atoms; gray spheres are fluorine atoms. The arrows indicate C3 and C4, respectively.
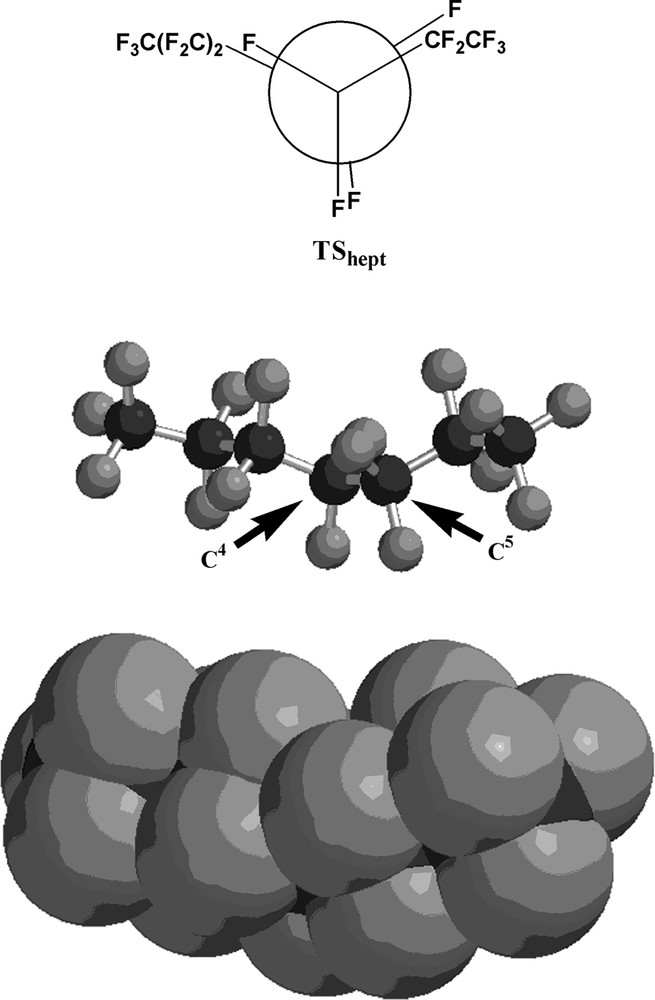
The transition state structure (TShept) for the interconversion of the two lowest-energy conformers (Ahex and Bhex) of perfluoroheptane. Black spheres are carbon atoms; gray spheres are fluorine atoms. The arrows indicate C4 and C5, respectively.
In order to confirm the presence of a transition state, vibrational analyses of both TShex and TShept were carried out. For the perfluorohexane transition state, only one imaginary vibrational frequency was found at 37.51 cm–1, and for the perfluoroheptane transition state, only one imaginary vibrational frequency was found at 36.56 cm–1. In each case the imaginary frequency corresponded to a slight torsional motion about the C–C bond carrying the eclipsed atoms and groups. Vibrational analyses on the ground state conformers Ahex, Bhex, Ahept, and Bhept exhibited no imaginary frequencies, as expected.
The experimental desorption energies of perfluorohexane and perfluoroheptane from a Pt(111) surface were 11.68 kcal mol–1 (48.87 kJ mol–1) and 12.86 kcal mol–1 (53.81 kJ mol–1) for the strongly adsorbed α1 species, respectively, while the less-strongly adsorbed α2 species desorbed with energies of 9.82 kcal mol–1 (41.09 kJ mol–1) and 10.75 kcal mol–1 (44.98 kJ mol–1), respectively [1]. In Ref. [1] it was assumed that the α1 species were staggered, zig–zag alkane-like conformers adsorbing to the surface, and these experimental values led to the conclusion that since perfluorohexane and perfluoroheptane differ by exactly one CF2 group, the adsorption energy per CF2 group was about 1.18 kcal mol–1 (4.94 kJ mol–1). Once this value was determined, the difference in desorption energies of the α1 and α2 species was seen to be equivalent to the loss of approximately one CF2 surface interaction for each molecule.
Recent reports demonstrating that the lowest-energy conformer of a perfluoroalkane is helical rather than a staggered, zig-zag alkane-like conformer alter the above analysis only slightly. Because of the helical structure of conformers A, only about five of the fluorine atoms of perfluorohexane and six of the fluorine atoms of perfluoroheptane can interact strongly with the Lewis acidic Pt metal surface. (If the lowest-energy conformers were the staggered, zig-zag alkane-like conformers, six fluorine atoms in perfluorohexane and eight fluorine atoms in perfluoroheptane could interact with the metal surface.) However, conformers B differ from A only in that the ‘kink’ in the perfluorocarbon chain reduces the surface interaction by about one fluorine atom; conformers B interact with a metal surface through four fluorine atoms in perfluorohexane and five in perfluoroheptane. Thus, the interaction energy per CF group is about 1.18 kcal mol–1 (4.94 kJ mol–1), rather than per CF2 group.
Based upon previous work by Sexton and Hughes [12] and Madey and Yates [13], the adsorption of perfluorocarbons on metal surfaces may be described by Eq. (1),(1)
The activation energies to attain the transition state for the interconversion of conformers A and B for perfluorohexane and perfluoroheptane are accessible (13.247 and 15.279 kJ mol–1) at the temperatures of the adsorption/desorption experiments (ca. 100 K); increased surface coverages and the crowding that results can provide the necessary activation energies to convert conformer A to conformer B for either perfluorocarbon. Further, the ‘kinked’ conformers B are visibly shorter than conformers A (for perfluorohexane, the longest end-to-end distance for Ahex is 8.527 Å, while for Bhex the longest end-to-end distance for the molecule is 7.781 Å; the analogous distances for perfluoroheptane are 9.797 and 9.007 Å, respectively). If adsorbed to the surface either directly from the gas phase or if formed on the surface due to crowding, the B conformers are likely to take up less space on the surface than will conformers A. Thus, again, increased surface coverages are likely to cause the conversion of conformers A to B, and to actually favor conformers B at higher coverages. Finally, it might be argued that some of the differences in adsorption arise from adsorbate–adsorbate interactions. However, since the B conformers are shorter than the A conformers, these possible adsorbate–adsorbate interactions will also be diminished because of the smaller size of the B conformers.
4 Conclusions
The stability of helical structures for the perfluorocarbons indicates that it is the helical structure that adsorbs to a platinum(111) surface at low sub-monolayer coverages (the α1 adsorbate). With increasing coverages of the perfluorocarbon, some of the α1 adsorbate is crowded into an interconversion to the α2 adsorbate, now identified as the next lowest-energy conformer of the perfluoroalkane. The energy differences between the two conformers for the two perfluoroalkanes studied, as well as the energy required to interconvert them (through the eclipsed transition state), are readily accessible at the temperatures of the experiment.
Acknowledgements
The authors wish to acknowledge the National Science Foundation's Course, Curriculum, and Laboratory Improvement Program, Adaptation and Implementation Section (NSF-9950344) for generous support in the purchase of computers and computational chemistry software. Matching funds and other support have been received from the Humboldt State University Department of Chemistry, the College of Natural Resources and Sciences, the Office for Research and Graduate Studies, and the Office of the Vice-President for Academic Affairs. This generous support, as well as support from the Humboldt State University Foundation, is gratefully acknowledged.