

1 Introduction
Selective signaling of heavy metal ions is a very important topic for the detection and treatment of the toxic metal ions in various chemical systems including living systems. In this field, calixarenes are currently the subjects of study as chemical sensors [1,2] and selective receptors [3] due to their important functionalization possibilities.
Then this led us to the development of a new class of chromoionophore receptors. We have chosen to graft on the upper rim of calix [4]arene phenylazo groups as chromophore and chelating agents on the lower rim.
Here we report the synthesis of derivatized azocalix [4]arenes bearing ester [4], amide [5], bipyridyl [6] and bithiazoyl [7] moieties. The extraction properties of this new class of azocalixarenes towards various cations will be presented. Moreover, in order to obtain more efficiency and selectivity in metal extraction properties of calixarenes, β-ketoimine groups have been appended on the lower rim of calixarenes.
2 Syntheses of calix [4]arene derivatives
2.1 Synthesis of ester/amide phenylazocalix [4]arene derivatives
The preparation of two bis(azo)calix [4]arenes 2 and 3 incorporating both ester and amide functions is described (Fig. 1). In order to graft phenylazo groups on the upper rim of calix [4]arenes and ester or/and glycine units on the lower rim, providing additional donor atoms [8], we have developed a strategy in three steps which permit to obtain these ligands. The first intermediate 26,28-dihydroxy-25,27-di(ethoxycarbonylmetoxy)calix [4]arene [9], which is a key reactant for the formation of 5,17-bis(phenylazo)-26,28-dihydroxy-25,27-di(ethoxycarbonylmethoxy) calix [4]arene (2) was chosen in order to block the two distal hydroxy groups of calix [4]arene. Thus using conventional copulation procedure [10], 2 can be obtained, respectively, in 52% yield. The tetra-substituted calix [4]arene 3 was obtained by reaction of 2 with the N-(chloroacetyl) glycine ethylester in presence of Na2CO3 as base and a catalytic amount of sodium iodide in acetonitrile at reflux temperature for 72 h in 33% yield. The cone conformation of compounds 2 and 3 was reflected in the characteristic AB system for the methylene groups bridging the aromatic rings in the 1H and 13C NMR spectrum [11].

Ester/amide phenylazocalix [4]arene derivatives.
The syntheses of tetrakis(phenylazo)calixarene derivatives 4, 5a and 5b were performed through a classic O-substitution on azocalixarene. The reaction of p-tetrakis(phenylazo)calix [4]arene [12] with tertiary acetamide (α-chloro-N,N-diethylacetamide) and Cs2CO3 in dry DMF gave compound 5b in an 1,3-alternate conformation. The analogous reaction in the presence of CaH2 as base gave 5a in a cone conformation. In this case, we can see that Ca2+ cation is capable of maintaining the cone conformation whereas for the synthesis of 5b [13], Cs+ permit to obtain an 1,3-alternate conformation in accordance with Refs. [14,15].
The reaction of p-tetrakis(phenylazo)calix [4]arene with α-chloro-N,N-diethylacetamide, CaH2 in DMF gave mainly the distal 1,3-disubstituted compound 4. The 1H and 13C NMR spectra indicate a cone conformation of compound 4. A similar reaction with an excess of K2CO3 gave potassium complex of 4, identified 4-K. This complex was isolated by chromatography column as crystal suitable for X-ray structure showing a cone conformation as depicted later.
2.2 Synthesis of heterocyclic phenylazocalix [4]arene derivatives
6-Bromomethyl-6′-methyl-2,2′-bipyridine [15] and 4-bromomethyl-4-methyl-2,2′-bithiazole [16] were synthesized according to the literature and grafted on the lower rim of phenylazocalix [4]arenes. The incorporation of only two phenylazo groups on the lower rim of calix [4]arene require to graft in first time the biheterocycle moieties. Then 25,27-di[(6-(6′-methyl-2,2′-bipyridyl)-yl)methoxy]-26,28-di(hydroxy)calix [4]arene and 25,27-di[(4-methyl-2,2′-bithiazolyl-4′-yl)methoxy]-26,28-di(hydroxy)calix [4]arene were synthesized [17] and chosen here in order to block the two distal hydroxy groups of calix [4]arene. Thus using conventional copulation procedure with substituted diazonium BF4– [10], 6 and 7 (Fig. 2) were obtained in cone conformation with 66% and 61%, respectively, yields.

Bithiazolyl and bipyridyl phenylazocalix [4]arene derivatives.
The reaction of p-tetrakis(phenylazo)calix [4]arene with 4-bromomethyl-4-methyl-2,2′-bithiazole, BaO, Ba(OH)2, 8H2O and 4-bromomethyl-4-methyl-2,2′-bithiazole in anhydrous DMF has resulted in the formation of compound 8 (64% yield) and the same reaction with 6-bromomethyl-6′-methyl-2,2′-bipyridine in anhydrous DMF has resulted in the formation of compound 9 (48% yield).
The diazo-coupling reaction of thiacalix [4]arene with substituted diazonium BF4 salt produced, as described in the literature, the p-tetrakis(phenylazo)-substituted this calix [4]arenes [18]. We attempted the preparation of dialkyl derivatives 11 by direct O-substitution of phenylazothiacalixarene with 4-bromomethyl-4-methyl-2,2′-bithiazole using various molar ratios between the azothiacalixarene and the base (Na2CO3). We found that using an excess of 4-bromomethyl-4-methyl-2,2′-bithiazole and a slight excess of Na2CO3 in anhydrous acetone, the azothiacalixarene was mainly di-O-substituted to give 11 as the major product (26% yield). Similarly, the 6-bromomethyl-6′-methyl-2,2′-bipyridine subunits was grafted on the lower rim of the azothiacalixarene platform using 5 eq. of Na2CO3 for gave compound 10 (18% yield).
2.3 Synthesis of di(β-ketoimine)calix [4]arene
β-Ketoimine calix [4]arene derivative has been prepared in three steps by the following reaction sequence as depicted in Fig. 3. In first time, nitrile functions were incorporated by O-substitution on the lower rim of calix [4]arene in distal position. The treatment of calix [4]arene with bromoacetonitrile and K2CO3 in dry acetonitrile gave the di-substituted nitrile compound 13. In second step, this compound was reduced into the corresponding amine 14 by classic procedure. Then, the amine calixarene derivative was condensated with pentan-2,4-dione in a mixture of ethanol/dichloromethane. Molecular sieves were used in order to trap water molecules. These reactions lead us to the desired di(β-ketoimine)calix [4]arene derivative 15.
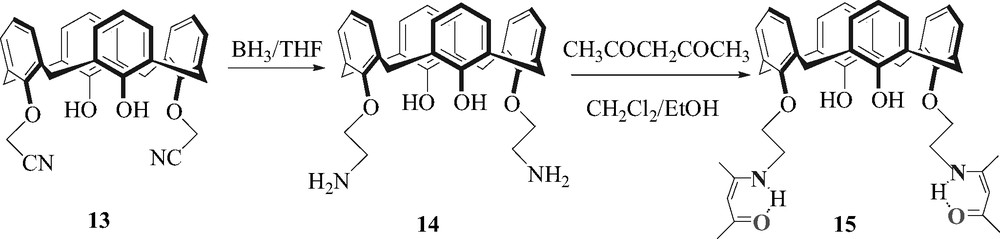
Reaction sequence for the synthesis of 25,27-bis-[(ß-ketoimine)-ethoxy]-26,28-dihydroxycalix [4]arene (15).
3 Extraction properties
3.1 Extraction properties of 1–5b
The results given in Fig. 4 correspond to the percentages of extracted cations (%E) from solutions of calixarene derivatives 2–5b. Concerning the diphenylazocalix [4]arene 1, no extraction of the tested cations was observed. The ester and ester/amide calixarene derivatives 2 and 3 show a significant extraction level for Na+, K+ and Ca2+ at pH 4.8 but have no affinity for the transition metals. The compound 3 is slightly more efficient to extract these alkali cations certainly due to the presence of more donor atoms on the lower rim of calixarene. All the amide calixarene derivatives 4, 5a and 5b show a significant extraction level only for sodium, potassium and calcium, K+ being the best extracted. This is coherent to the results obtained for similar compounds bearing tert-butyl groups at the upper rim [19]. The compound 4 presents a good efficiency of the extraction of potassium. This result correlates with the potassium complex 4-K that we have isolated and where the potassium is co-ordinated with the four oxygen atoms of the hydroxyl groups, two oxygen atoms of the amide substituents and one of the solvent (ethyl acetate) (Fig. 5).

Extraction percentages of calixarenes 2–5b at pH 4.8.
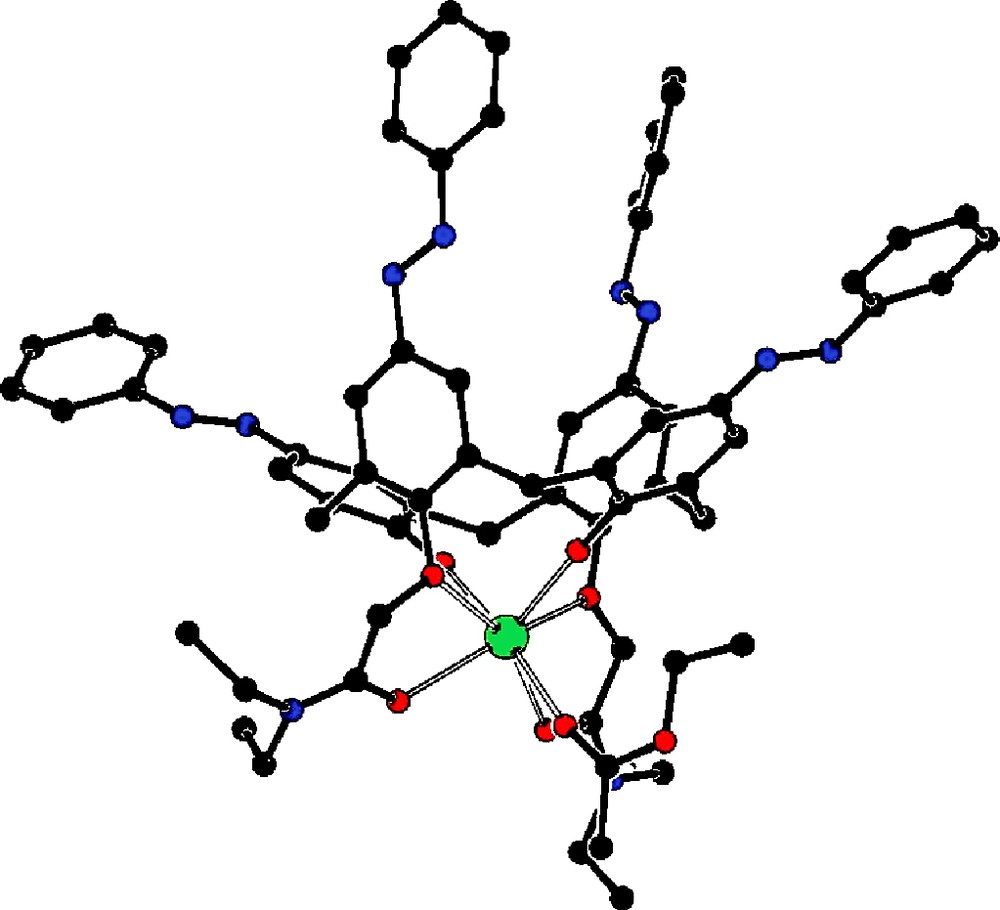
X-ray structure of complex 4-K.
The tetra-amide derivative 5a (cone conformation) is the most efficient for the extraction of Na+ (51%), K+ (75%) and Ca2+ (48%). In the case of compound 5b (1,3-alternate conformation), the extraction percents are close to those of 5a, 52% for K+ and 42% for Ca2+ but very low for Na+ (4%) and Mn2+ (4%). The di-amide derivative 4 extracts the same metals than 5b and 5a but exhibits a significant difference. This difference in the extraction behavior is attributable to the number of amide grafted on the calixarene. These results confirmed previous studies which have shown that calixarene amide derivatives are efficient ligands for alkali and alkaline earth metal ions [20].
These calixarenes are not efficient for the extraction of transition metals at pH values 4.8 and 2.2. Then we have modified the pH. The results show that in basic media (pH 8), only the compounds 2 and 3 are efficient and selective towards Pb2+ (74%).
3.2 Extraction properties of 6–12
In order to extract transition metals, the biheterocyclic calixarene and thiacalixarene derivatives were studied. The percentages of extraction of transition metal ions (Cu2+, Ag+, Pb2+ and Ni2+) at pH 5.3 were calculated (Fig. 6).
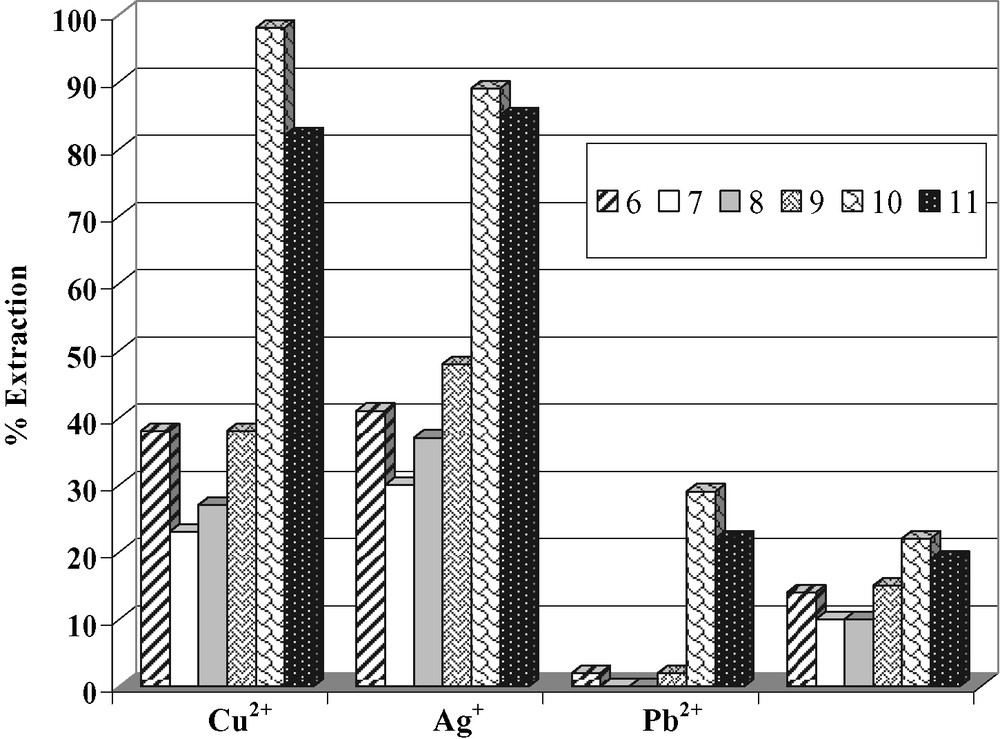
Extraction percentages of and heterocyclic azocalix [4]arenes 6–11 at pH 5.3.
First, we can see that the thiacalixarene 11 and 10 extract approximately two to three more than the corresponding calixarene 8 and 9. The thiacalix [4]arene derivatives 10 and 11 have an extraction ability with all the tested metal ions whereas calixarene derivatives 6–9 have no affinity for Pd2+ and extract slightly Ni2+ (around 10%). It is clear that the extractibility of Pb2+ by 10 and 11 arises from the sulfur bridges in thiacalixarene. All the compounds show more affinity toward Ag+ and Cu2+. The thiacalixarene 10 and 11 extract more than 80% of Cu2+ and Ag+. These results are probably due to the presence of the bis-heterocyclic subunits which have a high affinity with Cu2+ and Ag+ [21,22]. In all case, we can observe that the compounds bearing bipyridine 6, 9 and 10 are more efficient than the compounds incorporating bithiazoyl subunits 7, 8 and 11. We can think the extraction abilities of the bipyridyl moieties are higher than the one of bithiazolyl. We can also see that the replacement of methylene bridges in calix [4]arene by epithio groups affects the extraction ability.
Among these compounds, thiacalixarene derivatives present the best efficiency for the extraction of Cu2+ and Ag+.
3.3 Extraction properties of 15
The extraction data with (β-ketoimine)ethoxy calix [4]arene derivative 15 has been investigated for various cations and the extractibilities data have been collected in Fig. 7.
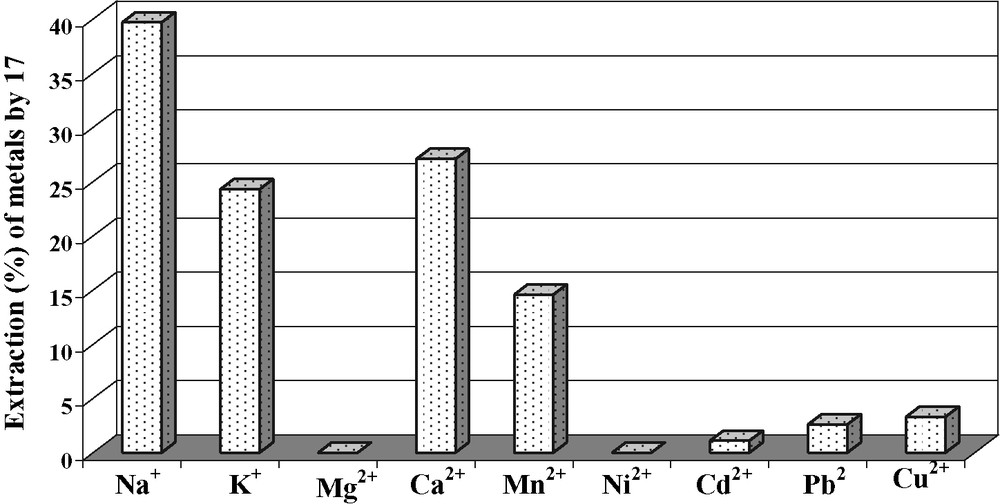
Extraction percentages of cations by 15 at pH 4.8.
The results of extraction show that this podand presents a good affinity for Na+ (39.7%), K+ (24.3%) and Ca2+ (27.1%). This is probably due to the electrostatic interaction between the cations and the cavity formed by the β-ketoimine groups. This compound does not extract the transition metals at pH values 4.8 and 2.2 and does not exceed 5% of extraction, except for Mn2+ (14.6%) at pH 4.8.
On the other hand, in a basic medium (pH 8) in the presence of pyridine as a synergistic agent, the compound 15 is an effective extractant for Pb2+ (86.7%) but it is ineffective for Cu2+, Ni2+ and Cd2+. These extractibilities seem to correlate with size effects of cations. Indeed, lead has the largest ionic radius of the three metals tested. Then these results encouraged us to study the selectivity of Pb2+ towards Ni2+, Cd2+ and Cu2+ in the same experimental conditions: A solution of four metal mixture (Pb2+, Ni2+, Cd2+ and Cu2+) at the same concentration (10–4 mol l–1) has been prepared. Our data show that 15 presents a very good selectivity for Pb2+ (84.8%) in comparison with Ni2+ (2.4%), Cd2+ (3.8%) and Cu2+ (2.8%) (Fig. 8). The presence of hydrogen substituents introduces the possibility of both intramolecular and intermolecular hydrogen bonding, which might influence the behavior of extraction.
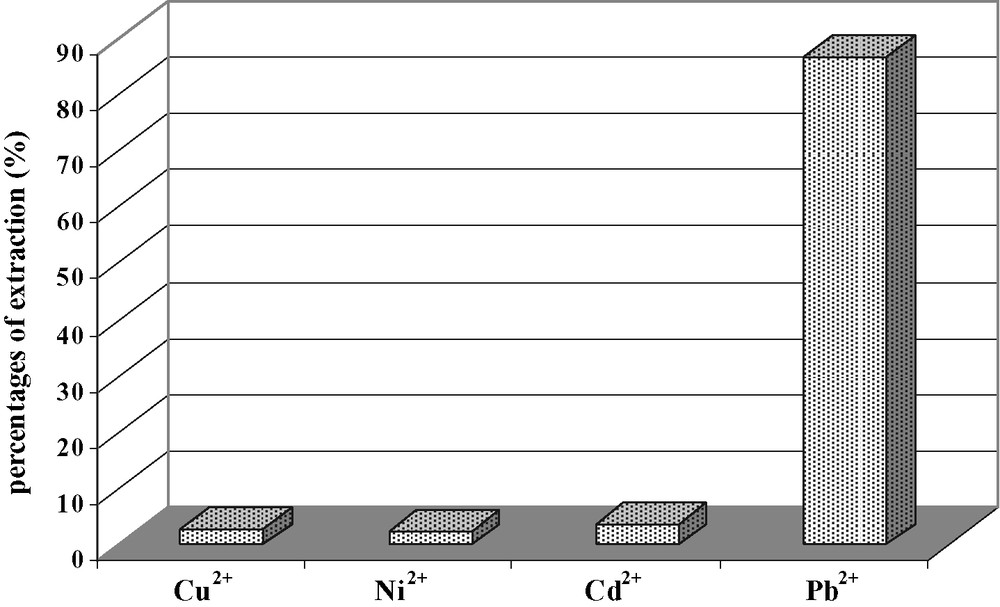
Extraction selective of Pb2+ by compound 15 at pH 8.
At our state of knowledge of calix [4]arene extraction with heavy metals, it is difficult to make any statements about the reasons for this observed selectivity. The receptor 15 exhibits a selectivity for Pb2+.
4 Conclusion
This paper describes the extraction properties of chromoionophore receptors bearing different chelating agents on the lower rim of azocalixarene and azothiacalixarene. This study show that the phenylazo groups on calixarene does not affect the extraction properties. The di-substituted β-ketoimine calixarene is an effective and selective extractant of Pb2+. The biheterocyclic moieties grafted on calixarenes promote the extraction of Cu2+ and Ag+. Among the described compounds, thiacalixarenes are better candidates than calixarenes for the extraction of transition metals.
5 Experimental
5.1 General remarks
Melting points were determined with an Electrothermal 9100 Capillary apparatus. 1H and 13C NMR spectra were recorded on Bruker DRX 300 spectrometer (1H: 300 MHz, 13C: 75 MHz; solvent, chemical shifts in ppm, J in Hertz). 13C/1H HMBC was recorded on Brucker AM 500. Mass spectra were obtained by electrospray technique, positive mode (ES-MS). Infrared was performed on a Mattson 5000 FT apparatus (nutilde in cm–1). Macherey–Nagel plates were used for TLC analyses (SiO2, Roth, polygram Sil G/UV 254) and silica gel 60 (Merck, particle size 0.040–0.063 mm) was used for chromatography columns. All solvents were purified by standard procedures before use. Dry solvents were obtained by literature methods and stored over molecular sieves. All other reagents grade quality as obtained from commercial suppliers were used without further purification. All reactions were carried out under nitrogen atmosphere. p-Tetrakis(phenylazo)tetrahydroxy-2,8,14,20-tetrathiacalix [4]arene was synthesized according to literature procedure [10]. Extraction studies: The extraction of metals was investigated using acetate and chloride salts. The organic solutions were made by dissolving a weighed amount of the compound in dichloromethane. The aqueous solution was buffered to pH 4.8 with tri(hydroxymethyl)aminomethane—HCl (0.05 M) (99%, Acros) or 5.3 with acetic acid (99% Acros) (1.8 × 10–3 M) and sodium acetate (99%, Fluka) (8.2 × 10–3 M). The ionic strength was maintained at μ = 0.1 with tetramethylammonium chloride or potassium chloride (0.1 M) (98%, Acros). Liquid–liquid extraction experiments were carried out in a flask by shaking for 12 h in a thermostated bath (30 °C), 25 ml of an aqueous phase containing metal salt (10–4 M) and 5 ml of organic phase containing the calixarene (5 × 10–4 M). The aqueous phase was separated, centrifuged, then 1% of HNO3 was added to these solutions and analyzed by atomic absorption spectrometry (Perkin Elmer 3110) with an air-acetylene flame, the measurements being carried out using standard conditions calibration. The percentages of extraction (Ex%) were determined from the equation [23] (1).(1)
5.2 Synthesis of 5,17-bis(phenylazo)-26,28-dihydroxy-25,27-di(ethoxycarbonylmethoxy) calix [4]arene (2)
25,27-Di(ethoxycarbonylmethoxy)-26,28(di-hydroxy)calix [4]arene (2 g, 3.37 mmol) and the BF4– salt benzenediazoniums (8.42 mmol) were dissolved in THF (100 ml). The reaction was initiated by the addition of pyridine (5 ml) to the cooled THF solution at 0 °C. After 48 h the mixture was evaporated to dryness. The residue was dissolved in a minimum of pyridine. The addition of methanol has given an orange precipitate which was recovered by filtration and washed with methanol. 2 was purified by chromatography column on silica gel (ethyl acetate/hexane; 4:6 (v/v)) to yield 2 as an orange solid (1.4 g, 52%). M.p.: 248–249 °C. 1H NMR (CDCl3) δ = 8.29 (s, 2H, OH), 7.85 (d, 4H, J = 7.17 Hz, H-diazo), 7.75 (s, 4H, H-diazo), 7.50–7.39 (m, 6H, H-diazo), 7.02 (d, 4H, J = 7.53 Hz, HAr), 6.78 (t, 2H, J = 7.53 Hz, HAr), 4.74 (s, 4H, OCH2CO), 4.53, 3.57 (‘q', AB, JAB = 13.17 Hz, ArCH2Ar), 4.37 (q, 4H, J = 7.14 Hz, OCH2CH3), 1.36 (t, 6H, J = 7.17 Hz, OCH2CH3). 13C NMR (CDCl3) δ = 169.16 (CO), 156.93, 153.43, 152.65, 146.09, 132.91, 130.36, 130.02, 129.40, 128.87, 126.30, 124.30, 122.76 (CAr), 72.90 (OCH2CO), 61.99 (OCH2CH3), 31.87 (ArCH2Ar), 14.58 (OCH2CH3). ES-MS m/z: 805.3 [M + H]+ (Calcd. 805.9), 827.3 [M + Na]+ (Calcd. 827.9). IR: 3381.7 (OH), 2981.5 (CAr–H), 2929.6 (CH2, CH3), 1749.9 (CO), 1473.1, 1461.3, 1441.5 (C=C, N=N).
5.3 Synthesis of 5,17-bis(phenylazo)-26,28-bis{[(ethoxycarbonyl)methylcarbamoyl]methoxy-25,27-di(ethoxycarbonylmethoxy) calix [4]arene (3)
A mixture of 2 (0.25 mmol), anhydrous sodium carbonate (0.265 g, 2.5 mmol), N-(chloroacetyl) glycine ethylester (1.75 mmol) and sodium iodide (catalytic amount) in acetonitrile (35 ml) were refluxed for 72 h. The solvent was then evaporated and the residue was taken up in CH2Cl2 (30 ml), washed with a saturated aqueous ammonium chloride solution (2 × 15 ml) and water (2 × 15 ml). After evaporation of the solvent the product was triturated with MeOH and filtered off and dried to give pure 3 as a red solid (0.09 g, 33%). M.p.: 92–94 °C. 1H NMR (CDCl3): 8.28 (t, 2H, J = 6.24 Hz, HNCH2), 7.86 (d, 4H, J = 7.14 Hz, H-diazo), 7.64 (s, 4H, HAr), 7.46–7.43 (m, 6H, H-diazo), 6.47 (t, 2H, J = 6.21 Hz, HAr), 6.40 (d, 4H, J = 6.78 Hz, HAr), 4.92 (s, 4H, OCH2CO), 4.77, 3.51 (‘q', AB, JAB = 14.13 Hz, ArCH2Ar), 4.54 (s, 4H, OCH2CO), 4.29–4.16 (m, 12H, HNCH2CO and OCH2CH3), 1.32 (t, 6H, J = 7.17 Hz, OCH2CH3), 1.30 (t, 6H, J = 6.96 Hz, OCH2CH3). 13C NMR (CDCl3): 171.09, 170.00, 169.89 (CO), 160.68, 154.98, 153.07, 148.48, 135.93, 132.85, 131.07, 129,44, 129.39, 124.62, 123.05 (CAr), 74.48, 72.91 (OCH2CO), 61.84, 61.61 (OCH2CH3), 40.94, 31.76 (ArCH2Ar), 14.55 (OCH2CH3). ES-MS m/z: 1091.2 [M + H]+ (Calcd. 1092.19), 1113.3 [M + Na]+ (Calcd. 1114.17). IR: 3395.3 (NH), 2964, 2932.8 (C–H), 1741.7, 1680.3 (CO), 1583.4, 1531.9, 1461.7, 1434.3 (HAr, C=C).
5.4 Synthesis of 5,11,17,23-tetrakis(phenylazo)-25,27-dihydroxy-26,28-bis[(N,N-diethylaminocar bonyl)methoxy] calix [4]arene (4)
p-Tetrakis(phenylazo)calix [4]arene (0.3 g, 0.36 mmol) and CaH2 (0.14 g, 3.3 mmol) in dry DMF (20 ml) were stirred for 2 h at 70 °C. After cooling to room temperature α-chloro-N,N-diethylacetamide (0.1 ml, 0.72 mmol) was added. The mixture was stirred for 22 h at 80 °C. To this solution was added water (80 ml). The resulting precipitate was filtered off and treated with 1 M HCl (35 ml) and CHCl3 (40 ml). The organic phase was separated, washed with water (20 ml), dried with MgSO4. After evaporation to dryness, purification of the residue by recrystallization from acetone gave 4 as a red powder (0.12 g, 31%). M.p. 187–189 °C. IR: 3345.8 (OH), 2971.8, 2930.2 (CH), 1655.2 (C=O), 1584.3, 1471.8, 1444.8 (C=C, N=N). 1H NMR (C5D5N): 1.15 (t, J = 7.1 Hz, 12H, NCH2CH3), 3.39 (q, J = 7.0 Hz, 4H, NCH2CH3), 3.51 (q, J = 7.2 Hz, 4H, NCH2CH3), 5.14, 3.85 (‘q', AX, JAX = 13.2 Hz, 8H, ArCH2Ar), 5.29 (s, 4H, CH2O), 7.67–7.19 (m, 28 H, HAr), 10.15 (br s, 2H, OH). 13C NMR (C5D5N): 12.8 (NCH2CH3), 13.9 (NCH2CH3), 32.0 (ArCH2Ar), 40.4 (NCH2CH3), 40.9 (NCH2CH3), 73.6 (CH2O), 122.7, 123.3, 124.6, 124.63, 128.9, 134.9, 135.3, 135.6 (CHAr), 146.0, 149.3, 149.6, 150.0, 151.2, 152.6, 153.1, 157.5 (CAr), 167.8 (CO). ES-MS: m/z = 1067.3 [M + H]+ (Calcd. 1067.49), 1089.2 [M + Na]+ (Calcd. 1089.48), 1105.3 [M + K]+ (Calcd. 1105.59).). Elemental analysis calcd. (%) for C64H62N10O6: C 72.03, H 5.86, N 13.12; found C 71.96, H 5.83, N 13.05.
5.5 Synthesis of 5,11,17,23-tetrakis(phenylazo)-25,26,27,28-tetra[(N,N-diethylaminocarbonyl) methoxy]calix [4]arene (5a) – cone conformation
p-Tetrakis(phenylazo)calix [4]arene (0.3 g, 0.36 mmol) and CaH2 (0.23 g, 6.6 mmol) were stirred under nitrogen in dry DMF (20 ml) at 70 °C for 2 h. After cooling to room temperature, α-chloro-N,N-diethylacetamide (0.3 ml, 8.64 mmol) was added to this solution. The mixture was stirred for 48 h at 80 °C then water (40 ml) was added. The resulting precipitate was recovered by filtration. The residue was dissolved in CHCl3 (40 ml) and 1 M HCl (35 ml). The organic phase was separated, washed with water (2 × 20 ml) and dried over MgSO4. After evaporation to dryness, purification of the compound by recrystallization from acetone gave 5a as a yellow–orange powder (0.08 g, 17%). M.p. 186–188 °C. IR: 2970.8, 2931.3 (CH), 1653.5 (CO), 1581.4, 1459.5, 1431,1 (C=C, N=N). 1H NMR (CDCl3): 1.16 (t, J = 7.1 Hz, 12H, CH3CH2N), 1.23 (t, J = 7.0 Hz, 12H, CH3CH2N), 3.43–3.34 (m, 16H, CH2N), 5.13 (s, 8H, CH2O), 3.56, 5.56 (‘q’, AX, JAX = 13.5 Hz, 8H, ArCH2Ar), 7.32–7.30 (m, 12H, Hm,p-diazo), 7.43 (s, 8H, HAr), 7.68 (d, J = 7.8 Hz, 8H, Ho-diazo). 13C NMR (CDCl3): 13.5 (NCH2CH3), 14.7 (NCH2CH3), 32.6 (ArCH2Ar), 40.5 (NCH2CH3), 41.3 (NCH2CH3), 72.3 (CH2O), 122.9, 124.1, 129.2, 130.3 (CHAr), 135.9, 148.8, 153.2, 159.9 (CAr), 168.6 (CO). ES-MS: m/z = 1293.5 [M + H]+ (Calcd. 1293.65). Elemental analysis calcd. (%) for C76H84N12O8: C 70.57, H 6.55, N 12.99; found C 70.31, H 6.55, N 12.99.
5.6 Synthesis of 5,11,17,23-tetrakis(phenylazo)-25,26,27,28-tetra[(N,N-diethylaminocarbonyl)methoxy] calix [4]arene (5b) – 1,3-alternate conformation
p-Tetrakis(phenylazo)calix [4]arene (0.3 g, 0.36 mmol) and Cs2CO3 (1.408 g, 4.32 mmol) were stirred under nitrogen in dry DMF (30 ml) at reflux temperature for 2 h. To this solution, α-chloro-N,N-diethylacetamide (8.64 mmol) was added. After 3 days, the reaction mixture was cooled to room temperature and 50 ml of water were added. The orange precipitate was recovered by filtration and washed three times with water (3 × 20 ml). The residue was dissolved in CHCl3 (30 ml) and 10% HCl (35 ml). The organic phase was separated and dried over Na2SO4. After evaporation to dryness, a red product was obtained and purified by chromatography column [SiO2, ethyl acetate/hexane/acetonitrile/methanol = 5:4.4:0.4:0.2 (v/v)] gave 5b as a red solid (0.042 g, 16%). M.p. 214–216 °C. IR: 2971, 2932, 2878 (CH), 1640.6 (C=O), 1598.0, 1450.0 (N=N, C=C). 1H NMR (CDCl3): 0.62 (t, J = 7.0 Hz, 12H, NCH2–CH3), 1.13 (t, J = 7.1 Hz, 12H, NCH2–CH3), 3.06 (q, J = 7.0 Hz, 8H, NCH2CH3) 3.37 (q, , J = 7.0 Hz, 8H, –NCH2CH3), 4.16 (s, 8H, ArCH2Ar), 4.44 (s, 8H, CH2O), 7.44–7.51 (m, 12H, Hm,p-diazo), 7.75 (s, 8H, HAr), 7.83 (d, J = 7.8 Hz, 8H, Ho-diazo).13C NMR (CDCl3): 13.3 (NCH2CH3), 14.4 (NCH2CH3), 38.0 (ArCH2Ar), 41.2, 42.7 (NCH2CH3), 72.3 (CH2O), 122.8, 125.4, 129.5, 131.0 (CHAr), 134.8, 147.9, 152.9, 161.7 (CAr), 167.7 (CO). ES-MS: m/z = 1293.5 [M + H]+ (Calcd. 1293.65). Elemental analysis calcd. (%) for C76H84N12O8 : C 70.57, H 6.55, N 12.99; found C 70.77, H 6.61, N 13.00.
5.7 Synthesis of 5,17-di(phenylazo)-25,27-di[(6-(6′-methyl-2,2′-bipyridyl)-yl)methoxy]-26,28-di(hydroxy)calix [4]arène (6)
A mixture of 25,27-[(4-méthyl-2,2′-bithiazolyl-4′-yl)méthoxy]26,28-bis(hydroxy) calix [4]arène (0.22 g, 0.28 mmol) and the BF4– salt benzenediazonium (0.84 mmol) were dissolved in THF (30 ml). The reaction was initiated by the addition of pyridine (1 ml) to the cooled THF solution at 0 °C. After 48 h, the orange precipitate was recovered by filtrated and washed with methanol. The product was recrystallized with CH2Cl2/MeOH (98:2) affording 6 as an orange powder (0.187 g, R = 66%). M.p.: 197–198 °C. 1H NMR (CDCl3): 2.66 (s, 6H, CH3-bpy); 3.46, 4.48 ('q', AB, JAB = 13.2, 8H, Ar–CH2–Ar); 5.28 (s, 4H, OCH2-bpy); 6.71–6.85 (m, 2H, H-bpy); 6.93–7.06 (m, 6H, H–Ar); 7.11–7.16 (m, 6H, H–Ar(azo)); 7.49–7.52 (m, 2H, H-bpy); 7.61–7.69 (m, 4H, H–Ar(azo)); 7.81 (m, 4H, H–Ar); 7.87 (d, J = 7.7, 2H, H-bpy); 8.07–8.15 (m, 2H, H-bpy); 8.23 (d, J = 7,6, 2H, H-bpy); 8.36 (d, J = 7,6, 2H, H-bpy); 8.65 (s, 2H, OH). 13C NMR (CD2Cl2): 24.68 (CH3-bpy); 32.13 (Ar–CH2–Ar); 79.71 (OCH2-bpy); 118.71, 121.44, 122.82, 122.88, 122.94, 123.78 (C(H)-bpy); 124.62, 129.40, 129.49, 130.70, 130.99, 138.57 (C(H)–Ar); 128.68, 128.97, 134.08, 137.44, 147.16, 148.05, 152.25, 152.97, 153.13, 154.63 (C–Ar, C-bpy). ES-MS m/z: 1003.3 [M + H]+ (calcd. 1003.15). IR: 3330 (OH); 3176 (Csp2–H); 2925.0 (Csp3–H); 1593 (C=N); 1523, 1471, 1444 (C=C); 1114 (C–O–C).
5.8 Synthesis of 5,17-di(phenylazo)-25,27-di[(4-methyl-2,2′-bithiazolyl-4′yl)methoxy]-26,28-di(hydroxy) calix [4]arene (7)
A mixture of calix [4]arene (0.229 g, 0.54 mmol) and K2CO3 (0.149 g, 1.07 mmol) were stirred in refluxing in anhydrous acetonitrile (40 ml) under nitrogen for 30 min. Then 0.296 g of 4-bromomethyl-4-methyl-2,2′-bithiazole (1.07 mmol) were added and this mixture was heated for 28 h. Then the solvent was removed and the residue was dissolved in CH2Cl2 (60 ml). The resulting solution was filtered off on Celite®. The filtrate was concentrated. A chromatopgrapy column on this residue (SiO2, CH2Cl2/MeOH, 99:1) gave 0.25 g (0.31 mmol) of 25,27-[(4-methyl-2,2′-bithiazolyl-4′-yl)methoxy]-26,28-bis(hydroxy)calix [4]arene as an intermediate. This one was then dissolved in 30 ml of THF. To this solution, 3 eq. of BF4– salt benzenediazonium were added. The mixture was stirred for 30 min at 0 °C. Upon the addition of 1 ml of pyridine, a red precipitate appears. After 48 h, the precipitate is filtered off and washed with methanol. The residue was recrystallized (CH2Cl2/MeOH, 98:2) to give 7 (0.197 g, 0.19 mmol, R = 61%): M.p.: 190–191 °C. 1H NMR ((CD3)2CO): 2.50 (s, 6H, CH3-btz); 3.92, 4.72 (‘q', AB, JAB = 13.3, 8H, Ar–CH2–Ar); 5.47 (s, 4H, OCH2-btz); 7.34 (s, 2H, H-btz); 7.38-7.49 (m, 6H, H–Ar); 7.70 (s, 4H, H–Ar); 7.75–7.77 (m, 6H, H–Ar(azo)); 7.85–7.92 (m, 4H, H–Ar(azo)); 8.03 (s, 2H, H-btz); 8.55 (s, 2H, OH). 13C NMR (CDCl3): 16.38 (CH3-btz); 32.53 (Ar–CH2–Ar); 72.55 (OCH2-btz); 122.93, 123.17 (C(H)-btz); 124.62, 129.34, 124.37, 129.42, 130.51, 131.25 (C(H)–Ar); 129.11, 135.70, 147.40, 147.62, 150.5, 153.27 (C–Ar); 153.62, 153.92, 157.20, 168.96 (C-btz). ES-MS m/z: 1043.0 [M + H]+ (calcd. 1043.2). IR: 3348 (OH); 1619 (C=N); 1376 (C–S); 1117.4 (C–O–C); 983, 882 (C–H btz).
5.9 Synthesis of 5,11,17,23-tetra(phenylazo)-25,27-di[(4-methyl-2,2′-bithiazolyl-4′-yl)methoxy]-26,28-di(hydroxy)calix [4]arene (8)
p-Tetrakis(phenylazo)calix [4]arene (0.113 g, 0.135 mmol), Ba(OH)2, 8H2O (0.127 g, 0.405 mmol) and BaO (0.062 g, 0.405 mmol) were mixed in dry DMF (5 ml) under nitrogen for 24 h at room temperature. After addition of 4-bromomethyl-4-methyl-2,2′-bithiazole (0.111 g, 0.405 mmol), the mixture was stirred at room temperature for 48 h. Then water (25 ml) was added to this solution and the resulting precipitate was filtered off, washed with water (25 ml) and then dissolved in CH2Cl2. The residue was extracted with water and dried over Na2SO4. The product was recrystallized with CH2Cl2/MeOH (99:1) affording 8 as red powder (0.073 g, 64%). M.p.: 245–246 °C. 1H NMR: 2.55 (s, 6H, CH3-btz); 3.91, 4.79 (‘q', AB, JAB = 13.5, 8H, Ar-CH2-Ar); 5.34 (s, 4H, OCH2-btz); 6.99 (s, 2H, H-btz); 7.45 (t, 8H, Hm-diazo); 7.53 (t, 4H, Hp-diazo); 7.71 (d, J = 8.7, 8H, Ho-diazo); 7.89 (s, 8H, ArH); 7.81 (s, 2H, OH); 8.32 (s, 2H, H-btz). 13C NMR: 17.15 (CH3-btz); 32.21 (Ar–CH2–Ar); 74.13 (OCH2-btz); 117.50, 120.04 (C(H)-btz); 123.37, 124.00, 124.32, 125.28, 129.27, 129.64, 134.37, 135.67, 136.00, 150.41, 154.85, 153.46 (CAr); 152.87, 153.52, 160.67, 162.77 (Cbtz). UV: 334 (83,400), 228 (66,800). ES-MS m/z: 1250.31 [M + Na]+ (calcd. 1250.6), 1229.31 [M + H]+ (calcd. 1229.5). Anal. calcd. for C68H52N12O4S4 (1228.31), C 66.43, H 4.26, N 13.67, O 5.21, S 10.43; found : C 66.48, H 4.32, N 13.61, O 5.28, S 10.39. IR: 3306 (stretching, OH), 3112–3054 (CH3-btz), 1593.7, 1543.1,1487.4, 1439.0 (C=C, N=N), 1420 (C–S), 975, 878 (btzC–H).
5.10 Synthesis of 5,11,17,23-tetra(phenylazo)-25,27-di[(6-(6′-methyl-2,2′-bipyridyl)-yl)methoxy]-26,28-di(hydroxy)calix [4]arene (9)
p-Tetrakis(phenylazo)calix [4]arene (0.350 g, 0.416 mmol), Ba(OH)2, 8H2O (0.391 g, 1.248 mmol) and BaO (0.190 g, 1.248 mmol) were stirred in anhydrous DMF (25 ml) under N2 for 2 h at ambient temperature. Then 0.328 g of 6-bromomethyl-6′-methyl-2,2′-bipyridine (1.248 mmol) were added and the mixture was stirred for 48 h. Upon the addition of water (40 ml) a precipitate appears. This one was filtered off, washed with water (20 ml) and dissolved in CH2Cl2. The organic phase was washed with water, separated and dried over Na2SO4. After evaporation to dryness, the compound was recrystallized in a mixture of CH2Cl2/MeOH (98:2) to give 9 as an orange powder (0.24 g, 0.2 mmol, R = 48%). M.p.: 243–244 °C. 1H-RMN (CDCl3): 2.66 (s, 6H, CH3-bpy); 3.76, 4.65 (‘q', AB, JAB = 13.2, 8H, Ar–CH2–Ar); 5.34 (s, 4H, OCH2-bpy); 7.15 (d, J = 7.7, 2H, H-bpy); 7.35–7.42 (m, 8H, H–Ar(azo)); 7.45 (d, J = 7.8, 2H, H-bpy); 7.50–7.69 (m, 8H, H–Ar(azo)); 7.65 (s, 4H, H–Ar); 7.72–7.75 (m, 4H, H-bpy); 7.91 (d, J = 7.3 4H, H–Ar(azo)); 7.93 (s, 4H, H–Ar); 8.23 (d, J = 7.7, 2H, H-bpy); 8.45 (d, J = 7.6, 2H, H-bpy); 8.58 (s, 2H, H–OH). 13C-RMN (CDCl3): 25.09 (CH3-bpy); 32.06 (Ar–CH2–Ar); 79.37 (OCH2-bpy); 118.73, 121.73, 122.86, 123.11, 137.43, 138.53 (C(H)-bpy); 123.87, 124.86, 128.17, 129.28, 129.44 (C(H)–Ar); 133.86, 146,30, 150.42, 152.90, 153.39, 154.67, 155.52, 155.76, 156.70, 157.12, 158.35 (C–Ar, C-bpy)). ES-MS m/z: 1205.4 [M + H]+ (calcd. 1205.3). IR: 3361 (elongation, OH); 3103 (Csp2–H); 2916 (Csp3–H); 1591 (C=N); 1508, 1472, 1445, 1408 (C=C); 1116 (C–O–C). Elemental analysis calcd. (%) for C76H60N12O4 (1204.4): C 75.73; H 5.02; N 13.94; O 5.31; found C 74.63; H 5.14; N 13.69; O 5.24.
5.11 Synthesis of 5,11,17,23-tetra(phenylazo)-25,27-di[(6-(6′-methyl-2,2′-bipiridyl)-yl)methoxy]-26,28-di(hydroxy)thiacalix [4]arène (10)
p-Tetrakis(phenylazo)tetrahydroxy-2,8,14,20-tetrathiacalix [4]arene (0.15 g, 0.16 mmol), Na2CO3 (0.084 g, 0.8 mmol) and 6-bromomethyl-6′-methyl-2,2′-bipyridine (0.33 g, 1.28 mmol) in refluxing acetone (30 ml) for 48 h. The product was purified by chromatography column on silica gel (CH2Cl2/petroleum ether/CH3CN; 50:74:26) to give red powder (0.071 g, 35%). Chromatography column on silica gel (ether de pétrole/AcOEt, 70:30) to give 10 as a red powder (0.038 g, 18%). M.p.: 278–279 °C. 1H NMR (C5D5N): 2.64 (s, 6H, CH3-bpy); 5.21 (s, 4H, OCH2-bpy); 7.10 (d, J = 7.6, 2H, H-bpy); 7.21–7.24 (m, 2H, H-bpy); 7.42–7.51 (m, 12H, H–Ar(azo)); 7.64 (m, 2H, H-bpy); 7.84–7.89 (m, 8H, H–Ar(azo)); 8.07 (m, 2H, H-bpy); 8.24 (s, 4H, H–Ar); 8.31 (d, J = 7.7, 2H, H-bpy); 8.45 (s, 4H, H–Ar)); 8.52 (d, J = 7.7, 2H, H-bpy). 13C NMR (C5D5N): 24.71 (CH3-bpy); 77.64 (OCH2-bpy); 122.69, 122.91, 123.11, 125.04, 125.33, 126.35 (C(H)-bpy); 129.37, 130.41, 133.65, 134.19 (C(H)–Ar); 134.80, 136.20, 147.95, 148.24, 148.81, 152.95, 153.45, 159.80, 167.18, 167.81, 168.91 (C–Ar, C-bpy). ES-MS m/z: 1277.4 [M + H]+ (calcd. 1277.5), 1300.1 [M + Na]+ (calcd. 1300.5). IR: 3361 (elongation, OH); 3104 (Csp2–H); 2922 (Csp3–H); 1508, 1472 (C=C); 1591 (C=N).
5.12 Synthesis of 5,11,17,23-tetra(phenylazo)-25,27-di[(4-methyl-2,2′-bithiazolyl-4′-yl) methoxy]-26,28-di(hydroxy)thiacalix [4]arene (11)
p-Tetrakis(phenylazo)tetrahydroxy-2,8,14,20-tetrathiacalix [4]arene (0.15 g, 0.16 mmol) and Na2CO3 (0.08 g, 0.8 mmol) were stirred in refluxing acetone (30 ml) under nitrogen for 1 h. After addition of 4-bromomethyl-4-methyl-2,2′-bithiazole (0.35 g, 1.28 mmol), the mixture was stirred at room temperature for 36 h. Then the solvent was removed and the residue was dissolved in CH2Cl2 (30 ml). The resulting solution was filtered off on Celite®. The filtrate was washed with HCl (0.1 N) and water (2 × 15 ml). The organic phase was dried over Na2SO4. After concentration, MeOH was added and the precipitated filtered off. Evaporation of the filtrate gave a raw material which was submitted to chromatography column on silica gel (ether de petrole/AcOEt, 70:30) to give 11 as a red powder (0.055 g, 26%). M.p.: 283–284 °C. 1H NMR (C5D5N): 2.55 (s, 6H, CH3-btz); 5.46 (s, 4H, OCH2-btz); 7.18 (s, 2H, H-btz); 7.32–7.43 (m, 12H, H–Ar(azo)); 7.75–7.81 (m, 8H, H–Ar (azo)); 7.98 (s, 4H, H–Ar); 8.24 (s, 2H, H-btz); 8.51 (m, 4H, H–Ar). 13C NMR (C5D5N): 17.19 (CH3-btz); 75.26 (OCH2-btz); 123.43, 123.35 (C(H)-btz); 125.18, 133.56, 135.36, 150.39 (C(H)–Ar); 124.01, 124.52, 134.98, 135.36, 135.98, 136.01, 149.67 (C–Ar); 144,65, 148.43, 156.45, 168.15 (C-btz). ES-MS m/z: 1302.1 [M + H]+ (calcd. 1302.6). IR: 3333 (stretching, OH); 3068 (CH3-btz); 1583, 1560, 1468, 1446 (C=C, N=N); 1404 (C–S); 905, 800 (btzC–H).
5.13 Synthesis of 25,27-di-(cyanomethoxy)-26,28-dihydroxycalix [4]arene (13)
A mixture of calix [4]arene (2 g, 4.7 mmol), K2CO3 (1.85 g, 13.4 mmol) and bromoacetonitrile (2.42 g, 20.2 mmol) in MeCN (30 ml) was stirred and heated under reflux for 7 h. Then the suspension was cooled to room temperature, filtered, and evaporated under reduced pressure. The product was purified by recrystallization (CHCl3/MeOH) to give 13 as a white powder (59%): M.p. 222–224 °C. 1H NMR (C5D5N) δ 3.62 (d, 4 H, J = 13.4 Hz), 4.59 (d, 4 H, J = 13.4 Hz), 5.26 (s, 4 H), 6.30 (t, 2 H, J = 7.5 Hz), 6.80 (d, 4 H, J = 7.5 Hz), 6.94 (t, 2 H, J = 7.5 Hz), 7.29 (d, 4 H, J = 7.5 Hz), 7.56 (s, 2 H). 13C NMR (C5D5N) δ 31.6, 61.0, 116.2, 120.0, 126.2, 128.5, 129.3, 129.7, 132.9, 151.4, 153.4. IR ν 3485, 3367 (OH), 3028, 2930 (C–H), 1592, 1466, 1456, 1444 (C=C). ES-MS m/z = 503.1 [M + H]+ (calcd. 503.6), 525.1 [M + Na]+ (calcd. 525.6), 541.1 [M + K]+ (calcd. 541.7).
5.14 Synthesis of 25,27-bis-(aminoethoxy)-26,28-dihydroxycalix [4]arene (14)
A solution of compound 13 (2.8 mmol) in 100 ml of dry THF was cooled using on ice-bath and then 60 ml (60 mmol) of BH3 (1 M in THF) was added dropwise under nitrogen. The reaction mixture was then heated at 80 °C for 8 h. After cooling, it was quenched with 50 ml of 1 N HCl and stirred for 1 h. The solvent was removed under reduced pressure and the residue was stirred with 50 ml of 6 N HCl and heated under reflux for 3 h. After being cooled, the acidic solution was washed with ether and then evaporated to dryness. The residue was suspended in dichloromethane and 2 N NaOH was added until basic pH. The organic layer was separated, dried and evaporated to give 14 (1.4 g, 98%). M.p. 186–188 °C. 1H RMN (C5D5N) δ 3.33 (t, 4 H, J = 4.7 Hz), 3.49 (d, 4 H, J = 13.0 Hz), 4.02 (t, 4 H, J = 4.7 Hz), 4.55 (d, 4 H, J = 12.8 Hz), 6.31 (t, 2 H, J = 7.5 Hz), 6.86 (d, 4 H, J = 7.7 Hz), 6.94 (t, 2 H, J = 7.3 Hz), 7.3 (d, 4 H, J = 7.3 Hz). 13C RMN (C5D5N) δ 31.8, 43.2, 79.5, 120.1, 124.0, 125.7, 129.7, 129.0, 133.9, 151.8, 154.0. IR ν 3238 (NH, OH), 2929, 2868 (C–H), 1593, 1465, 1456, 1432 (C=C). ES-SM m/z = 511.2 [M + H]+ (calcd. 511.25), 533.2 [M + Na]+ (calcd. 533.24).
5.15 Synthesis of 25,27-bis-[(ß-ketoimine)-ethoxy]-26,28-dihydroxycalix [4]arene (15)
1.2 mmol of 14, an excess of acetylacetone and molecular sieves are dispersed in an ethanol/dichloromethane mixture (3:1 v). The mixture is heated under reflux during 7 h under nitrogen. The mixture is filtered on dry silica and concentrate. The residue was purified by column chromatography on silica gel (EtOAc/hexane; 8:2, Rf = 0.56) and recrystallized in CH2Cl2/MeOH to give 15 (0.69 g, 85%). M.p. 232–234 °C. 1H RMN δ 1.92 (s, 6 H), 2.09 (s, 6 H), 3.40 (d, 4 H, J = 13.3 Hz), 3.83 (q, 4 H, J = 6.2 Hz), 4.07 (t, 4 H, J = 6.2 Hz), 4.24 (d, 4 H, J = 13.2 Hz), 5.01 (s, 2 H), 6.64 (t, 2 H, J = 7.5 Hz), 6.70 (t, 2 H, J = 7.5 Hz), 6.78 (d, 4 H, J = 7.5 Hz), 6.87 (s, 2 H), 7.09 (d, 4 H, J = 7.5 Hz), 11.01 (t, 2 H, J = 6.2 Hz). 13C RMN δ 19.4, 29.2, 31.4, 43.0, 75.8, 96.9, 119.5, 125.8, 129.0, 129.4, 128.4, 133.0, 152.1, 153.5, 163.4, 195.9. IR ν 3384 (NH, OH), 2921, 2879 (C-H), 1610 (CN), 1583, 1514, 1455 (C=C). ES-MS m/z = 674.9 [M + H]+ (calcd. 675.34), 697.2 [M + Na]+ (calcd. 697.33). C42H46N2O6 (674.3): calcd. C 74.75, H 6.87, N 4.15; found C 74.71, H 6.82, N 4.12.