

1 Introduction
Charge- (CT) and electron-transfer (ET) salts involving organic molecules, as well as coordination or organometallic compounds, as components of molecule-based conductors (e.g., [TTF][TCNQ] [1], K2[Pt(CN)4]X0.3·3H2O [2] and [TTF][M(dmit)2] (M = Ni, Pd) [3,4] superconductors (e.g. [TMTSF]2[ClO4] [5], RbCs2[C60] [6] and α-[EDT-TTF][Ni(dmit)2] [7]) and magnets (e.g. [TDAE][C60] [8] [FeCp*2][TCNE] [9] and [Mn(TPP)][TCNE] [10]) are a broad, rapidly expanding class of new materials that exhibit distinctive properties from those of the isolated molecules or ions [11].
Following the experimental observation that several homo- and hetero-metallic carbonyl clusters are multivalent or exhibit electrochemically reversible redox behaviour [12], we became interested in the properties of their salts with redox-active countercations, with the aim to contribute a new class of molecule-based charge- or electron-transfer salts belonging to the above categories of materials. The viologens (1,1′-disubstituted-4,4′-bipyridilium cations) appeared suitable candidates as redox-active countercations, because they feature 2+/1+ and 1+/0 redox changes with formal potentials (E°′) [13] comparable with those of several metal carbonyl clusters. Moreover, the E°′ of their redox changes is tunable as a function of the alkyl substituents and reaction solvents [13]. As a first attempt, we investigated the synthesis of the ethylviologen (EtV) salts of the [Ag13Fe8(CO)32]3–/4–/5– series of clusters [14,15]. The choice of the latter stems from the sufficiently good match between the EtV2+/EtV+• and [Ag13Fe8(CO)32]3–/[Ag13Fe8(CO)32]4–, as well as the EtV+•/EtV0 and [Ag13Fe8(CO)32]4–/[Ag13Fe8(CO)32]5– redox couples. Moreover, the odd-electron [Ag13Fe8(CO)32]4– anion, at difference from the even-electron [Ag13Fe8(CO)32]3–/5– species, displays an unambiguous EPR spectrum both in solution and in the solid state, which may be considered an additional fingerprint to its νCO IR absorptions [14].
As a result, we have isolated and characterised the missing [Ag13Fe8(CO)32]5– pentaanion in its ethylviologen radical monocation (EtV+•) salt. A comparison of the individual molecular parameters in the [Ag13Fe8(CO)32]3–/4–/5– series of clusters enables experimental quantomechanic considerations, which are in qualitative agreement with EH molecular orbital calculations. A further reason of interest derives from the packing of ethylviologen radical cations, which points out the probable presence of weak intercationic interactions. No previous example of structural characterisation of a salt involving viologens and a metal carbonyl cluster has been reported in the literature, the only related system being to our knowledge the methylviologen salt of [Pt12(CO)24]2– employed in catalysis [16,17].
2 Results and discussion
2.1 Reaction of [Ag13Fe8(CO)32]3–/4– with EtV2+2I– and synthesis of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF
On investigating the reaction of the [Ag13Fe8(CO)32]3–/4– salts with 1,1′-diethyl-4,4′-bipyridilium diiodide (EtV2+2I–), the possible effect on redox equilibrium constants of the solvents used during the working up should not be overlooked. Owing to the presence in literature of scattered data, determination of the redox changes of both EtV2+/+/0 and [Ag13Fe8(CO)32]3–/4–/5– under constant experimental conditions in the miscellaneous solvents has been unavoidable. Our data well compare with those previously reported and the most relevant are collected in Table 1.
Formal electrode potentials (mV) vs. SCE of [Ag13Fe8(CO)32]n– (n = 3, 4, 5) and EtVn+ (n = 2, 1, 0) a
| Redox couple | DMF | MeCN |
| EtV2+/EtV•+ | –405 | –390 |
| EtV•+/EtV0 | –790 | –820 |
| [Ag13Fe8(CO)32]3–/[Ag13Fe8(CO)32]4– | –310 | –370 b |
| [Ag13Fe8(CO)32]4–/[Ag13Fe8(CO)32]5– | –700 | –650 b |
a Pt working electrode, [NBu4]BF4 5 × 10–2 M supporting electrolyte, T = 298 K, 8.33 mV s–1 scan rate, EtVI2 and [NBu4]4[Ag13Fe8(CO)32] 5 × 10–4 M.
b Values from [15].
As inferable from Table 1, the EtV2+/EtV+• and [Ag13Fe8(CO)32]3–/[Ag13Fe8(CO)32]4– redox couples display so close formal potentials (E°′) in dimethylformamide (DMF) and MeCN that equilibrium constants of ca. 10–1 may be expected for reaction (1). Nevertheless, addition of a DMF solution of EtV2+ 2I– to a corresponding solution of [NBu4]4[Ag13Fe8(CO)32] failed to confirm the above expectation.(1)
Only decomposition to Ag metal, AgI, Fe(CO)5, [HFe3(CO)11]– and [Fe(CO)4I]– could be monitored. In no instance, IR absorptions attributable to [Ag13Fe8(CO)32]3– were observed. The decomposition is accompanied by a colour change of the reaction solution from red–brown to blue, that is clearly indicative of the formation of the EtV+• radical cation. Its progressive formation at the expenses of the odd-electron [Ag13Fe8(CO)32]4– anion is confirmed by IR and EPR monitoring. The latter unambiguously shows the typical fingerprints of both EtV+• and [Ag13Fe8(CO)32]4–. It seems reasonable to suggest that the [Ag13Fe8(CO)32]4– tetraanion is not simply oxidised by EtV2+ according to equilibrium reaction (1), owing to concomitant or subsequent reaction of the Ag–Fe cluster with the iodide counterions of EtV2+. These lead to degradation of the cluster into Ag metal, AgI, [Fe(CO)4I]– and other iron carbonyl side products. The relevance of the presence of iodide ions in the above degradation is in keeping with the results of analogous attempts to obtain ethylviologen salts of the [Fe3Pt3(CO)15]2–/–/0 series of clusters, which partially led to isolation of EtV2+/EtV+• salts of homometallic Pt carbonyl clusters via elimination of [Fe(CO)4I]– [18]. The IR pattern of [Fe(CO)4I]– (νCO at 2021 (mw) and 1916 (s) cm–1) is coincident with that of a genuine sample and its nature has been unequivocally confirmed by ESI-MS experiments.
Completely similar results have been obtained by treatment of preformed tetrasubstituted ammonium salts of [Ag13Fe8(CO)32]3– with EtV2+ 2I– in MeCN.
In the attempt to intercept some [Ag13Fe8(CO)32]n– (n = 3–5) as EtV2+ and/or EtV+• salt, the sodium salt of [Ag5Fe4(CO)16]3– has been prepared by reaction of AgNO3 with Na2[Fe(CO)4]·xTHF in MeCN according to Ref. [19], and treated with EtV2+ 2I–. Stepwise addition to the above solution of the EtV2+ iodide leads to progressive formation of a dark precipitate and a red solution containing Fe(CO)5, [HFe3(CO)11]– and [Ag13Fe8(CO)32]4–. Such a result is probably due to occurrence in solution of reaction (2), a lesser extent of reaction (3) and some degradation.(2)
According to formal potential of the EtV2+/EtV+• and [Ag13Fe8(CO)32]4–/[Ag13Fe8(CO)32]5– redox couples (Table 1), reaction (3) should display an equilibrium constant of ca. 105, both in MeCN and DMF. Its occurrence in the above experiments is probably limited by the adopted ca. 1:1 ratio between [Ag5Fe4(CO)16]3– and EtV2+, as well as precipitation of [Ag13Fe8(CO)32]5– as EtV+• salt. The low solubility of this salt also limits degradation by iodide ions.
The dark precipitate resulting from reaction (2) is insoluble in most organic solvents but soluble in DMF. The colour of the DMF solution is deep green. Since the [Ag13Fe8(CO)32]5–, as well as [Ag13Fe8(CO)32]4–, is red–brown and EtV•+ is blue, the green colour might be indicative of occurrence in solution of ion-pairing and charge–transfer. In partial agreement with this conclusion, monitoring of the above DMF solution by EPR only discloses the presence of the EtV+• radical monocation. Very weak features due to [Ag13Fe8(CO)32]4– only show up at the highest intensity gains. In apparent contrast, the IR spectrum of the DMF solution shows carbonyl absorptions at 1976 (s) and 1890 (m) cm–1, which are practically coincident with those of the odd-electron [Ag13Fe8(CO)32]4– anion in its [NBu4]+ salt. This apparent incongruence may find explaination in depletion of charge from [Ag13Fe8(CO)32]5– because of charge–transfer to EtV+• and consequent decreased back-donation to CO. Notice that reaction (4) should display an equilibrium constant of ca. 10–2 and 10–3, respectively, in DMF and MeCN.(4)
The dark-green precipitate has been successfully crystallised by layering isopropyl alcohol on top of the above DMF solution (2:1 in volume). The resulting crystals are brown and completely insoluble in all solvents, DMF included. Elemental analysis and EPR investigations of the crystals (Fig. 1a, b) are in keeping with the [EtV+•]5[Ag13Fe8(CO)32]·4 DMF formula resulting from X-ray diffraction analysis (Section 2.2). As shown in Fig. 1a, the EPR spectrum of the crystals only displays the signal of the EtV+• radical cation. A signal attributable to the odd-electron [Ag13Fe8(CO)32]4– anion shows up (Fig. 1b) only at very high gain. On assuming that no spin-pairing occurs because of inter-cation interactions (see Sections 2.2 and 2.3), signals integration would suggest an approximate 1:182 ratio between [Ag13Fe8(CO)32]4– anions and EtV+• radical cations. This would mean a ca. 1:35 ratio between [Ag13Fe8(CO)32]4– and [Ag13Fe8(CO)32]5– ions. In case the inter-cation interactions inferable from the X-ray structure (which points out the presence of [EtV]55+• pentameric aggregates), are sufficiently significant to give rise to the partial spin pairing suggested by EHMO calculations, the above ratio could reduce to ca. 1:181. The presence of [Ag13Fe8(CO)32]4– paramagnetic ions can be due to its faulty association in the [EtV+•]5[Ag13Fe8(CO)32]·4 DMF crystal lattice. However, in view of the calculated value of the equilibrium constant of reaction (4), the observed amount of [Ag13Fe8(CO)32]4– might also be in keeping with an electron-transfer process in the solid state, such as that represented by Eq. (4).
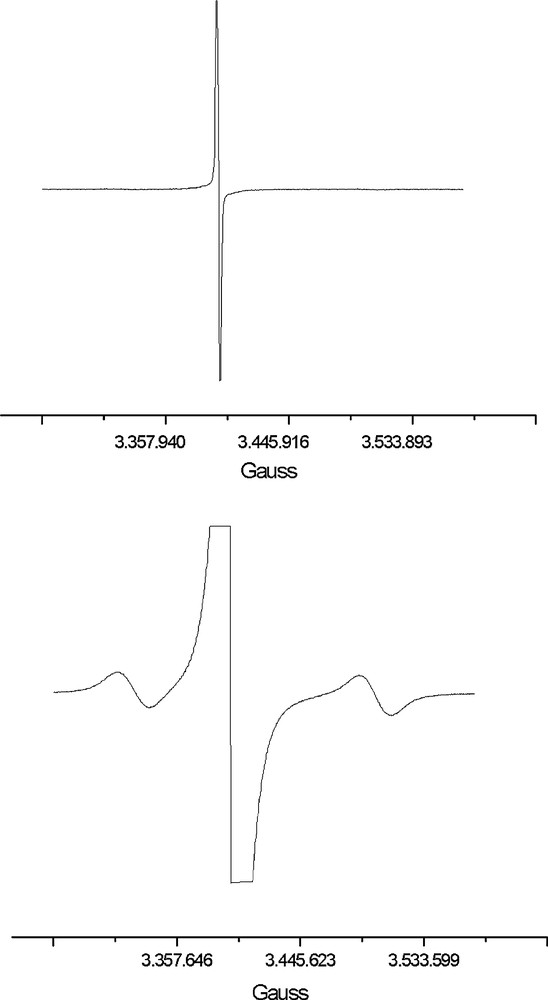
Solid-state EPR spectra at different gain intensity of a polycrystalline sample of [EtV•+]5[Ag13Fe8(CO)32]·4 DMF.
2.2 X-ray structure of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF
The unit cell of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF contains two [Ag13Fe8(CO)32]5– half-anions, four complete EtV+• cations and two half-cations and four DMF molecules. Most significant bond lengths of anion and cations are collected in Table 2, while the structure of [Ag13Fe8(CO)32]5– is shown in Fig. 2. The numbering scheme of the metal framework is reported in Fig. 3. As shown in Fig. 2, the metal core of the [Ag12(μ12-Ag)(μ3-Fe(CO)4)8]5– anion consists of a centred cuboctahedron of silver atoms with the triangular faces capped by iron atoms. The eight iron atoms individuate a non-bonded Fe8 cube. Each iron atom bears four carbonyl ligands and the μ3-Fe(CO)4 fragments adopt a C3v symmetry. The [Ag12(μ12-Ag)(μ3-Fe(CO)4)8]5– pentaanion is isostructural with the previously reported [Ag12(μ12-Ag)(μ3-Fe(CO)4)8]n– (n = 3, 4) anions [14,15]. However, due to some shrinking of the Ag13 cuboctahedron following a slight shortening of all Ag–Ag bonds and a comparable lengthening of Ag–Fe interactions, the Ag4Fe4 faces of the cube are noticeably more concave than for [Ag13Fe8(CO)32]n– (n = 3, 4). Centre–surface (range 2.871(1)–2.935(1) Å) and surface–surface (range 2.864(1)–2.964(1) Å) Ag–Ag interactions are very close and average 2.895 and 2.903 Å, respectively. Conversely, the Ag–Fe distances range between 2.729(2) and 2.766(2) Å and average 2.753 Å.
Most relevant bond lengths (Å) for [EtV•+]5[Ag13Fe8(CO)32]·4 DMF
| [Ag13Fe8(CO)32]5– | |||||
| Ag–Ag | |||||
| Ag(1)–Ag(7) | 2.871(1) | Ag(1)–Ag(7)#1 | 2.871(1) | Ag(1)–Ag(4)#1 | 2.883(1) |
| Ag(1)–Ag(4) | 2.883(1) | Ag(1)–Ag(6)#1 | 2.893(1) | Ag(1)–Ag(6) | 2.893(1) |
| Ag(1)–Ag(2) | 2.894(1) | Ag(1)–Ag(2)#1 | 2.894(1) | Ag(1)–Ag(3)#1 | 2.895(1) |
| Ag(1)–Ag(3) | 2.895(1) | Ag(1)–Ag(5) | 2.935(1) | Ag(1)–Ag(5)#1 | 2.935(1) |
| Ag(2)–Ag(5) | 2.873(1) | Ag(2)–Ag(3) | 2.881(1) | Ag(2)–Ag(4)#1 | 2.886(1) |
| Ag(2)–Ag(6)#1 | 2.964(1) | Ag(3)–Ag(5) | 2.865(2) | Ag(3)–Ag(4) | 2.906(1) |
| Ag(3)–Ag(7)#1 | 2.943(2) | Ag(4)–Ag(6) | 2.864(2) | Ag(4)–Ag(2)#1 | 2.886(2) |
| Ag(4)–Ag(7)#1 | 2.916(2) | Ag(5)–Ag(6) | 2.885(1) | Ag(5)–Ag(7) | 2.893(2) |
| Ag(6)–Ag(7) | 2.868(1) | Ag(6)–Ag(2)#1 | 2.964(2) | Ag(7)–Ag(4)#1 | 2.916(2) |
| Ag(7)–Ag(3)#1 | 2.943(2) | ||||
| Ag–Fe | |||||
| Ag(2)–Fe(9) | 2.748(2) | Ag(2)–Fe(10) | 2.749(2) | Ag(3)–Fe(8) | 2.729(2) |
| Ag(3)–Fe(10) | 2.765(2) | Ag(4)–Fe(8) | 2.748(2) | Ag(4)–Fe(9)#1 | 2.765(2) |
| Ag(5)–Fe(11) | 2.744(2) | Ag(5)–Fe(10) | 2.766(2) | Ag(6)–Fe(11) | 2.762(2) |
| Ag(6)–Fe(9)#1 | 2.767(2) | Ag(7)–Fe(8)#1 | 2.741(2) | Ag(7)–Fe(11) | 2.751(2) |
| Fe–C | |||||
| Fe(8)–C(3) | 1.734(17) | Fe(8)–C(2) | 1.75(2) | Fe(8)–C(1) | 1.767(17) |
| Fe(8)–C(4) | 1.779(17) | Fe(9)–C(8) | 1.733(19) | Fe(9)–C(5) | 1.736(16) |
| Fe(9)–C(7) | 1.753(17) | Fe(9)–C(6) | 1.79(2) | Fe(10)–C(9) | 1.752(17) |
| Fe(10)–C(12) | 1.779(17) | Fe(10)–C(10) | 1.780(17) | Fe(10)–C(11) | 1.784(17) |
| Fe(11)–C(16) | 1.743(18) | Fe(11)–C(13) | 1.755(18) | Fe(11)–C(15) | 1.76(2) |
| Fe(11)–C(14) | 1.799(16) | ||||
| C–O | |||||
| C(1)–O(1) | 1.192(16) | C(2)–O(2) | 1.201(19) | C(3)–O(3) | 1.161(17) |
| C(4)–O(4) | 1.137(16) | C(5)–O(5) | 1.148(16) | C(6)–O(6) | 1.14(2) |
| C(7)–O(7) | 1.179(16) | C(8)–O(8) | 1.171(17) | C(9)–O(9) | 1.131(16) |
| C(13)–O(13) | 1.146(18) | C(10)–O(10) | 1.169(16) | C(11)–O(11) | 1.144(16) |
| C(12)–O(12) | 1.153(16) | C(14)–O(14) | 1.133(16) | C(15)–O(15) | 1.165(19) |
| C(16)–O(16) | 1.172(17) | ||||
| EtV•+ | |||||
| N(1)–C(104) | 1.341(17) | N(1)–C(100) | 1.359(18) | N(1)–C(110) | 1.427(17) |
| C(100)–C(101) | 1.329(18) | C(101)–C(102) | 1.412(17) | C(102)–C(103) | 1.376(17) |
| C(102)–C(107) | 1.423(17) | C(103)–C(104) | 1.345(18) | ||
| N(2)–C(109) | 1.319(18) | N(2)–C(105) | 1.346(18) | N(2)–C(112) | 1.52(2) |
| C(105)–C(106) | 1.318(18) | C(106)–C(107) | 1.454(17) | C(107)–C(108) | 1.417(17) |
| C(108)–C(109) | 1.331(19) | C(110)–C(111) | 1.48(2) | ||
| N(3)–C(200) | 1.348(19) | N(3)–C(204) | 1.349(19) | N(3)–C(210) | 1.51(2) |
| C(200)–C(201) | 1.32(2) | C(201)–C(202) | 1.419(19) | C(202)–C(203) | 1.426(19) |
| C(202)–C(207) | 1.47(2) | C(203)–C(204) | 1.31(2) | ||
| N(4)–C(209) | 1.333(19) | N(4)–C(205) | 1.362(18) | N(4)–C(212) | 1.52(2) |
| C(205)–C(206) | 1.341(19) | C(206)–C(207) | 1.407(19) | C(207)–C(208) | 1.412(19) |
| C(208)–C(209) | 1.34(2) | C(210)–C(211) | 1.45(2) | C(212)–C(213) | 1.49(2) |
| N(5)–C(300) | 1.363(17) | N(5)–C(304) | 1.390(18) | N(5)–C(305) | 1.424(19) |
| C(300)–C(301) | 1.305(19) | C(301)–C(302) | 1.430(19) | C(302)–C(302)#2 | 1.42(3) |
| C(302)–C(303) | 1.435(19) | C(303)–C(304) | 1.29(2) | C(305)–C(306) | 1.53(2) |
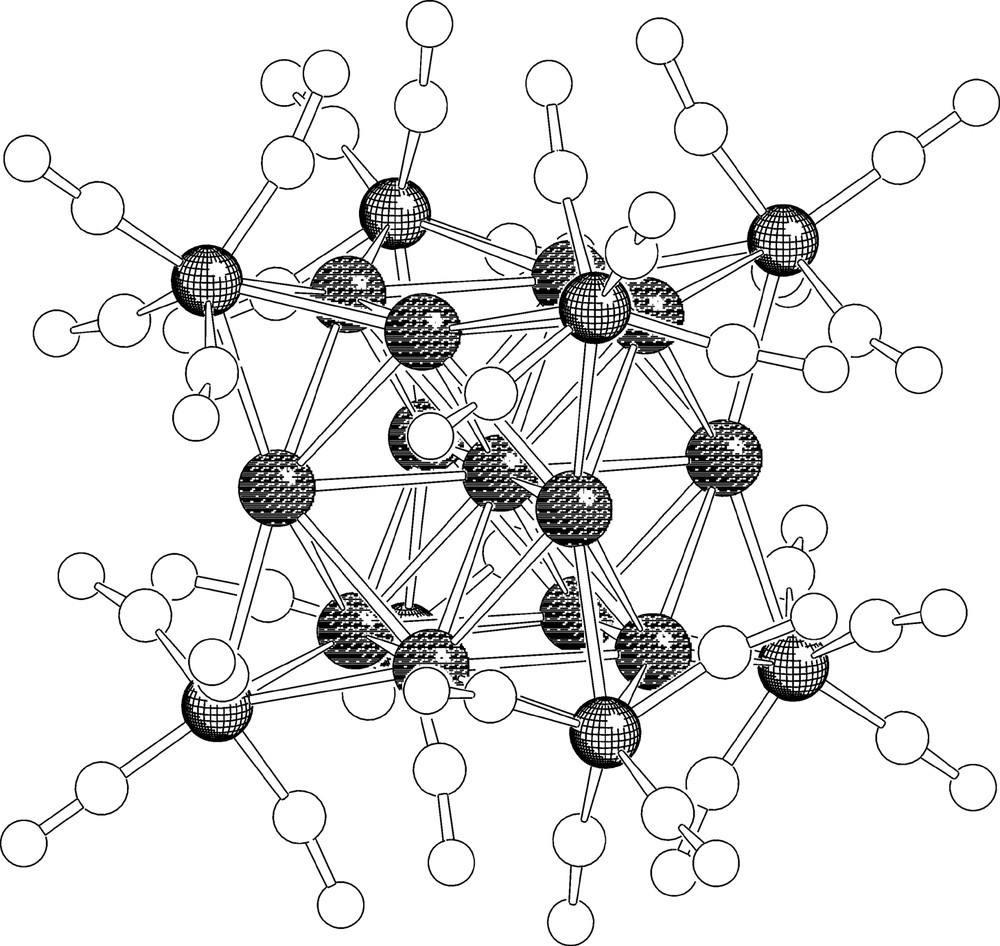
Structure of the [Ag13Fe8(CO)32]5– molecular ion.
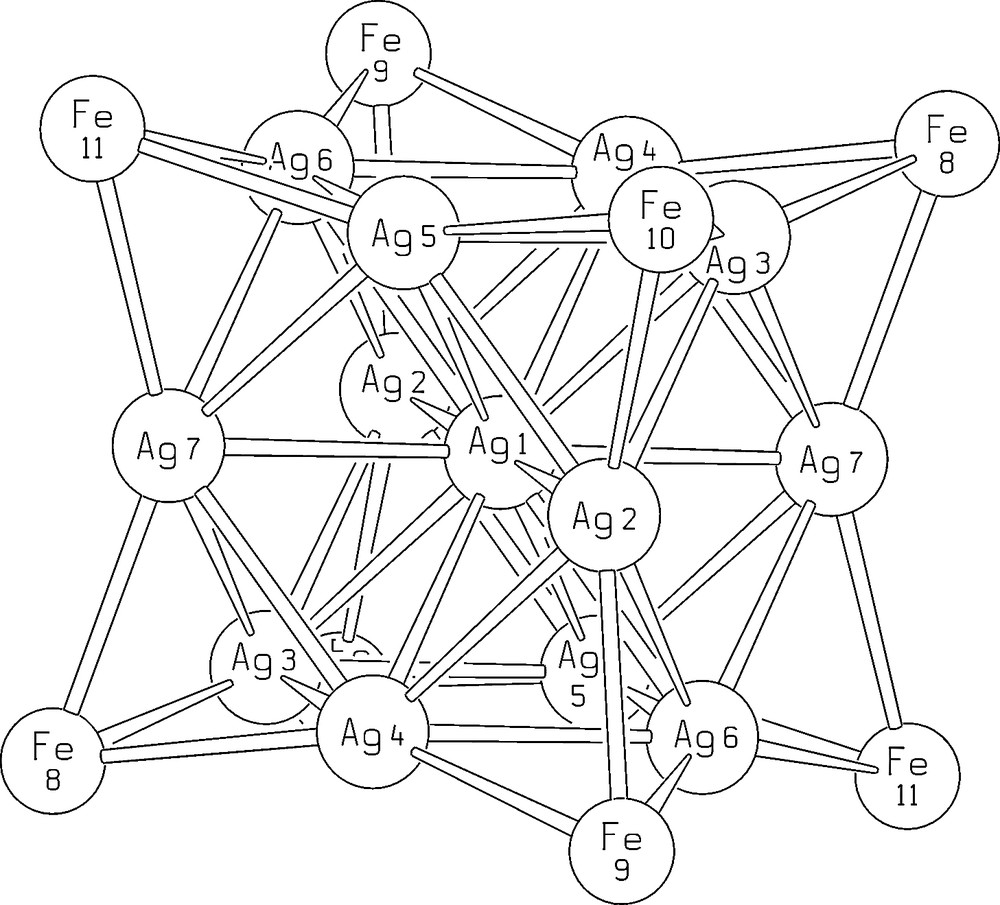
Numbering scheme for the metal frame of the [Ag13Fe8(CO)32]5– molecular ion.
The carbonyl ligands adopt a trigonal bipyramid arrangement around the Fe atom occupying one axial and three equatorial positions. The remaining axial position is taken by an Ag3 face of the cuboctahedron. The equatorial and axial CO ligands show different Fe–C, C–O distances and Fe–C–O angles: 1.79, 1.15, 164° and 1.74, 1.16, 179°, respectively. The bending of the equatorial carbonyls seems indicative of weak bonding interaction with the silver atoms (Ag······C range: 2.53(2)–2.70(2) Å).
The structure of the EtV+• countercations deserves some comments and their packing requires a detailed analysis. Several solid-state structures of 1,1′-dialkyl-4,4′-bipyridilium dications salts have been reported [13]. Analysis of the available structures brought to the conclusion that, in the absence of charge–transfer interactions with the anion, there is a non-zero dihedral angle between the two pyridil moieties. Dihedral angles up to 50° have been observed (e.g., the methylviologen salt of [PdCl4]2– [20]). The skewness is suggested to decrease down to 0°, as the strength of the CT interaction increases [13].
To the best of our knowledge, only one X-ray structure has so far been reported for a bipyridilium radical cation, namely the methylviologen [MeV+•][PF6–] salt [21]. This contains infinite stacks of MeV+• ions of two distinct types twisted by an angle of 37° and featuring a single interionic distance of 3.29 Å. The major differences between the two distinct MeV+• ions are represented by their interannular C–C distance (1.40 and 1.44 Å) and torsional angles (6 and 11°, respectively). This result is in agreement with conclusions drawn by ab initio calculations, which indicate that the radical cation, at difference from the skew (ca. 44.7°) dication, is almost flat (dihedral angle 11.9 or 13.6°) [22,23]. It has also been reported that MeV+• undergoes a monomer-dimer (as well as higher aggregates) equilibrium in water, which quenches the EPR signal [13].
The EPR of the crystals, the solid state stereochemistry of cations, and the stoichiometry of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF clearly point out that the 1,1′-diethyl-4.4′-bipyridilium moieties are EtV+• radical cations. Indeed, they are practically flat (skew angles are comprised in a 0–4° range) and their molecular parameters indicate an incipient non-aromatic, polyene-like structure, as a result of a small average shortening of the two facing intra- and inter-annular C–C bonds.
Besides, the EtV+• cations form pillars in which aggregates of five molecular ions displaying a repeating A–B–A′–B′–A sequence are clearly identifiable. Within each pentameric aggregate (see Fig. 4), the two outer units (A) are identical. The inner (A′) radical cation is twisted of ca. 23° with respect the A units and differs from the latter in showing a specular trans conformation of the methyl groups of the ethyl moieties, which are almost perpendicular to the bipyridilium plane. The B and B′ EtV+• cations are sandwiched by a A and A′ couples and are in a staggered conformation with respect to A, being twisted by ca. 90°. Both B and B′ display cis conformations of the methyl groups and are approximately related by an idealised mirror plane containing A′. Within each pentameric aggregate, there are interlayer non-bonding C....C contacts as short as 3.24 Å. In spite of the expected coulombic repulsions between EtV+• cationic moieties, the interplane separation (ca. 3.2 Å) is shorter than in graphite (3.35 Å). Along the pillars there are A·····A interlocks between the pentameric aggregates featuring interplane C·····N non-bonding distances of 3.67 Å as shortest contacts.
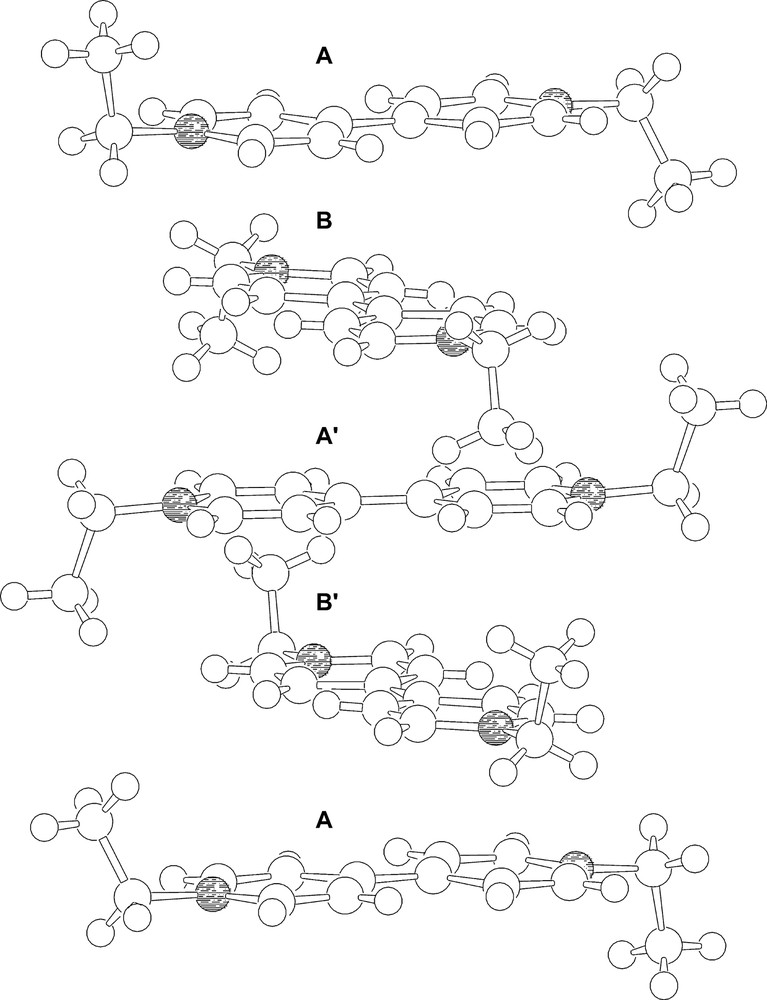
The aggregate of five EtV+• radical cations giving rise to infinite pillars.
2.3 EHMO calculations on [Ag13Fe8(CO)32]5– and EtV+• aggregates
It is worth commenting the nature of the HOMO of the [Ag13Fe8(CO)32]5– pentaanion since the observed structural variations in the [Ag13Fe8(CO)32]3–/4–/5– series (see Table 3) enable experimental quantomechanic [24]. Moreover, the EPR of [Ag13Fe8(CO)32]4– gives the unique possibility to experimentally test the reliability of any adopted theoretical method. First of all, the observed coupling constants of the unpaired electron with Ag, by comparison with isolated Ag atoms in noble gas matrices, points out a spin population of 0.25 on the interstitial Ag atom [14]. The [Ag13Fe8(CO)32]4– has already been investigated by EH and DFT methods [14,25]. EH calculations suggested that the singly-occupied molecular orbital (SOMO) is essentially non-bonding [14]. The structural variations reported in Table 3 prompted a more detailed analysis because of the small but significant shortening of Ag–Ag contacts on going from the tri- to the penta-anion, which is in contrast with the increasing free negative charge. As previously reported, the SOMO essentially derives from interaction of the 5 s atomic orbital of the interstitial Ag atom with a bonding and antibonding FMO of the Ag12Fe8(CO)32 cage. The 5 s atomic orbital of the interstitial Ag atom gives a 22% contribution to the SOMO, which is in an astonishingly good agreement with the experimental EPR results. Furthermore analysis of the overlap populations suggests a weak bonding character of the Aginner–Agouter and Agouter–Agouter interactions, a weak antibonding character of the Ag–Fe interactions and an essentially non-bonding character of the Fe–C and C–O bonds. The above suggestions are in agreement with experimental quantomechanic conclusions, which may be drawn from the trend of bonding contacts in the [Ag13Fe8(CO)32]3–/4–/5– series of compounds. Previous difficulties in observing such a trend are probably to ascribe to entanglement between two small and contrasting effects: the small swelling of the anion arising from the increasing free negative charge and comparable small shrinking of the Ag13 cuboctahedron due to the nature of the SOMO [15].
Comparison of average M–M and Fe–CO distances in the [Ag13Fe8(CO)32]n– (n = 3–5) anions
| Ag–Ag | Ag–Fe | Fe–Cax | Fe–Ceq | |
| [Ag13Fe8(CO)32]3– | 2.929 | 2.712 | 1.74 | 1.79 |
| [Ag13Fe8(CO)32]4– | 2.923 | 2.737 | 1.74 | 1.79 |
| [Ag13Fe8(CO)32]5– | 2.900 | 2.753 | 1.74 | 1.77 |
In consideration of the astonishingly good match between experimental and theoretical results for the [Ag13Fe8(CO)32]3–/4–/5– series of compounds, we also analysed the EHMO of the EtV+• monomer and the [EtV]55+• aggregate. The SOMO of the EtV+• monomer (Fig. 5a) falls 2.65 eV above the second occupied MO and 1.71 eV below the LUMO. It is bonding in character for the two facing intraring and the interannular C–C interactions and antibonding for all other C–C and C–N bonds within the rings. Therefore, its partial population causes the appearance of a polyene-like structure. Besides, according to EHMO calculations, the π-symmetry SOMO of the miscellaneous A, B, A′, B′, A units can weakly interact along the perpendicular direction to give rise to a narrow (0.52 eV wide) set of five weakly stabilised and destabilised σ-MOs (see Fig. 5b), which falls in the middle of a wide HOMO–LUMO gap (4.06 eV). The populated orbitals give rise to a very weak bonding interaction in the direction perpendicular to the EtV+• planes and can justify the occurrence of [EtV]55+• pentameric aggregates showing an A–B–A′–B′–A stack. That a twisted interlayer overlap of π clouds could be so effective to overcome coulombic repulsions among EtV+• radical cations is in agreement with X-ray findings. However, further experiments are necessary in order to establish if such an overlap is sufficient to cause partial electron-pairing, as suggested by EHMO calculations.
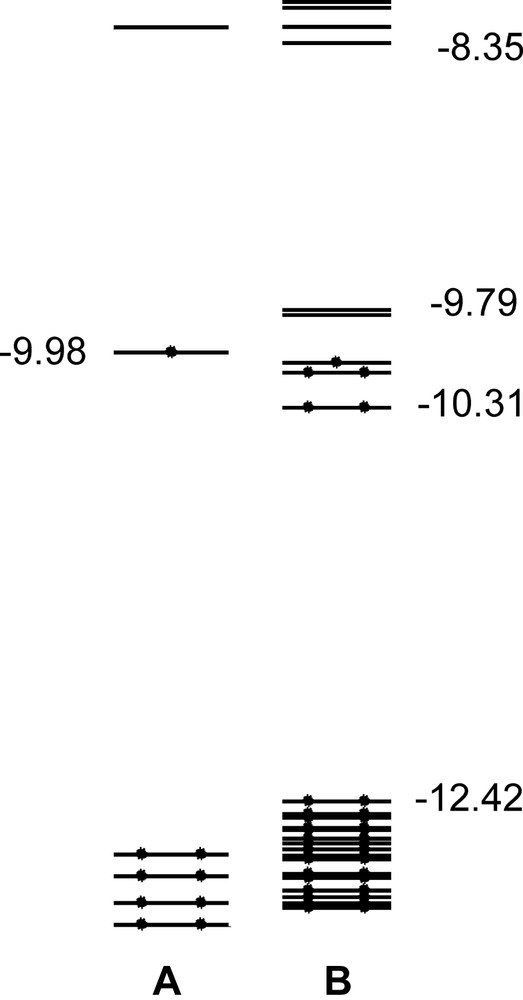
The frontier region (–13 and –8 eV range of energy) of an EtV+• radical cation (a) and the A–B–A′–B′–A [EtV]55+• aggregate (b), according to EHMO.
It is probably worthnoting that the presence of two alternating and distinct MeV+• ions along the infinite stacks of methylviologens of the [MeV+•][PF6–] salt has previously been interpreted in term of valence bond as a [MeV+• MeV+• ↔ MeV0 MeV2+] charge–transfer or self-complexation resonance [21]. By a related reasoning, a resonance of the kind [EtV+• EtV+• EtV+• EtV+• EtV+• ↔ EtV2+ EtV0 EtV+• EtV0 EtV2+] might be suggested in the present case. However, the individual molecular parameters of the A, A′, B and B′ units are only partially in agreement with the above charge distribution. For instance, the longest (1.47 Å) interannular C–C distance is found in A and would be compatible with its dicationic nature. In contrast the A′, B and B′ units display shorter and very similar contact interannular contacts of 1.42 Å (A′) and 1.43 Å (B and B′), which are not in keeping with their distinct nature of EtV+• and EtV0 species, respectively. It is necessary to stress that standard deviations of C–C and C–N distances of above EtV moieties are unfortunately too high to safely confirm or rule out such an interpretation.
2.4 Conclusions
In summary, the attempted preparation of EtV2+/EtV+• and [Ag13Fe8(CO)32]3–/[Ag13Fe8(CO)32]4– salts led to isolation of the missing [14,15] [Ag13Fe8(CO)32]5– pentaanion, the first X-ray structure of an ethylviologen radical cation and a snapshot of the interaction between viologen radical cations to give higher aggregates than dimers. On the basis of presently available data, it seems reasonable to suggest that [EtV+•]5[Ag13Fe8(CO)32] upon precipitation from MeCN solution gives rise to an amorphous green charge–transfer salt. Upon crystallisation from DMF, brown [EtV+•]5[Ag13Fe8(CO)32]·4 DMF crystals are obtained, in which EtV+• is present as [EtV]55+• pentamers. Its complete insolubility in all organic solvents (DMF included) prevented other spectroscopic studies than solid state EPR. Therefore, the purported CT behaviour and the relevance of the [EtV]55+• pentameric aggregates should be assessed by further physical characterisations. Anyway, the reported features of the title compound point out that viologen salts of redox-active metal carbonyl clusters could become promising molecule-based materials which, via fine tuning of their respective redox potentials, may assume potential interest as new CT and ET salts, as well as magnets.
3 Experimental
All reactions including sample manipulations were carried out with standard Schlenk techniques under nitrogen and in carefully dried solvents. The Na2[Fe(CO)4]·x THF (x = 1.5), Na3[Ag5Fe4(CO)16] and [NBu4]n[Ag13Fe8(CO)32] (n = 3, 4) salts were prepared according to literature [26,14,15,21]. Analyses of Fe and Ag have been performed by atomic absorption on a Pye-Unicam instrument. Infrared spectra were recorded on a Perkin Elmer 1605 interferometer using CaF2 cells. EPR experiments were carried out on a Bruker ER 041 XG instrument. Electrochemical measurements were carried out with a Radiometer PGP201 potentiostat. Potential values are referred to the saturated calomel electrode. EH calculations and structure drawings have been performed with CACAO98 [27] and SCHAKAL99 [28], respectively.
3.1 Synthesis of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF from Na3[Ag5Fe4(CO)16]
Solid AgNO3 (2.24 g, 13.18 mmol) was added in portions over a period of 2 h to a stirred suspension of Na2[Fe(CO)4]·xTHF (2.81 g, 8.73 mmol for x = 1.5) in MeCN (40 ml). The resulting brown suspension, which shows infrared carbonyl absorptions at 1965, 1948 and 1878 cm–1, was filtered and a MeCN solution of EtVI2 (1.19 g, 2.54 mmol) was added under stirring. The resulting red–brown suspension was filtered and the precipitate was thoroughly washed with THF and MeCN up to a colourless filtrate. The solid dark-green residue (55% yields based on silver) was dissolved in DMF (20 ml) and filtered. [EtV+•]5[Ag13Fe8(CO)32]·4 DMF separated out as well-shaped brown crystals from the DMF solution by layering isopropyl alcohol (40 ml). The crystals are insoluble in THF, alcohol, acetone, acetonitrile and DMF. [Found: Ag, 33.8; Fe, 10.6; C, 33.1; N, 4.6; H, 2.9. Calcd for [EtV+•]5[Ag13Fe8(CO)32]·4 DMF: Ag, 34.12; Fe,10.87; C, 33.32; N, 4.77; H, 2.89].
3.2 X-ray data collection and crystal structure determination of [EtV+•]5[Ag13Fe8(CO)32]·4 DMF
Crystal data and details of the data collection and refinement are given in Table 4. The diffraction experiments were carried out at room temperature on Bruker SMART2000 diffractometer equipped with a CCD detector. Intensity data were corrected for Lorentz and polarisation effects. An empirical absorption correction was applied by using SADABS [29]. The structure was solved by direct methods and refined by full-matrix least-squares on F2 using SHELXL97 [30]. The metal atom positions were determined by direct methods and all non-hydrogen atoms located from successive Fourier difference syntheses. Hydrogen atoms were added in calculated positions (dC–H 0.93 Å) and their positions were not refined but continuously updated with respect to their carbon atoms and were given a fixed isotropic thermal parameters.
Crystal data and structure refinement for [EtV•+]5[Ag13Fe8(CO)32]·4 DMF
| Empirical formula | C57H59Ag6.50Fe4N7O18 | |
| Formula weight | 2054.67 | |
| Temperature | 298(2) K | |
| Wavelength | 0.71073 Å | |
| Crystal system, space group triclinic | P | |
| Unit cell dimensions | a = 15.414(2) Å | α = 118.530(4)° |
| b = 16.674(3) Å | β = 101.387(4)° | |
| c = 16.743(3) Å | γ = 105.679(4)° | |
| Volume | 3357.9(9) Å3 | |
| Z, Calculated density | 2, 2.032 Mg m–3 | |
| Absorption coefficient | 2.759 mm–1 | |
| F(000) | 2007 | |
| Crystal size | 0.20 × 0.10 × 0.05 mm | |
| θ Range for data collection | 1.42–25.00° | |
| Limiting indices | –18 ≤ h ≤ 18, –19 ≤ k ≤ 19, –19 ≤ l ≤ 19 | |
| Reflections collected/unique | 30,108/11,816 [R(int) = 0.1433] | |
| Completeness to θ = 25.00 | 100.0% | |
| Maximum and minimum transmission | 0.8744 and 0.6084 | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 11816/32/835 | |
| Goodness-of-fit on F2 | 0.835 | |
| Final R indices [I > 2σ(I)] | R1 = 0.0649, wR2 = 0.1213 | |
| R indices (all data) | R1 = 0.1816, wR2 = 0.1473 | |
| Largest difference in peak and hole | 0.787 and –0.910 e Å–3 |
CCDC-236005 contains the supplementary crystallographic data for [EtV+•]5[Ag13Fe8(CO)32]·4 DMF. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (int.) +44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk].
Acknowledgements
We wish to acknowledge the financial contributions of the University of Bologna and MIUR (PRIN2003 and FIRB2002).