

1 Introduction
The work of the transition metal carbonyl clusters containing p-block elements has attracted considerable attention and extensively expanded because they exhibit unusual structural and reactivity patterns [1,2]. The previous studies in this area largely focused on the syntheses and structural features of the new clusters. However, the reactivities of the resultant clusters were less studied, and the effects of the main group elements on the cluster formation were particularly less explored. In this review, we will report the recent developments of the chalcogen-containing iron carbonyl clusters in our laboratory, dealing especially with the syntheses, reactivity comparison, and cluster growth and transformation processes of the smaller-sized clusters to the larger-sized ones.
As known, metal carbonyl clusters tend to fragment as applied in the catalytic reactions. However, compounds containing bonds between transition metals and group 16 elements (S, Se, Te) have been investigated [2] because of the potential usefulness of these main group elements as bridges between different metal atoms in clusters and as stabilizing ligands to prevent their fragmentation. In contrast to the larger number of sulfur containing clusters, those containing selenium and tellurium have been much less explored. Due to the relatively larger radius and Lewis acidity of selenium and tellurium, certain geometry and reactivity differences have been suggested for their resultant clusters. Nevertheless, the recent studies indicate that the electronic and steric factors of the incoming species may as well play the important role on the cluster formation. Such studies can reveal the valuable information regarding the selective anchoring process on metal surfaces that could lead to nanomaterials of controlled properties [3]. Hence, much more work has to be done to understand the complex effects on the cluster formation and transformations.
We will describe in this review mainly the structural transformations and cluster growth of the chalcogen-containing triiron clusters [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) toward a series of organic and inorganic electrophiles basically in terms of the effects of chalcogen atoms and the incoming electrophiles.
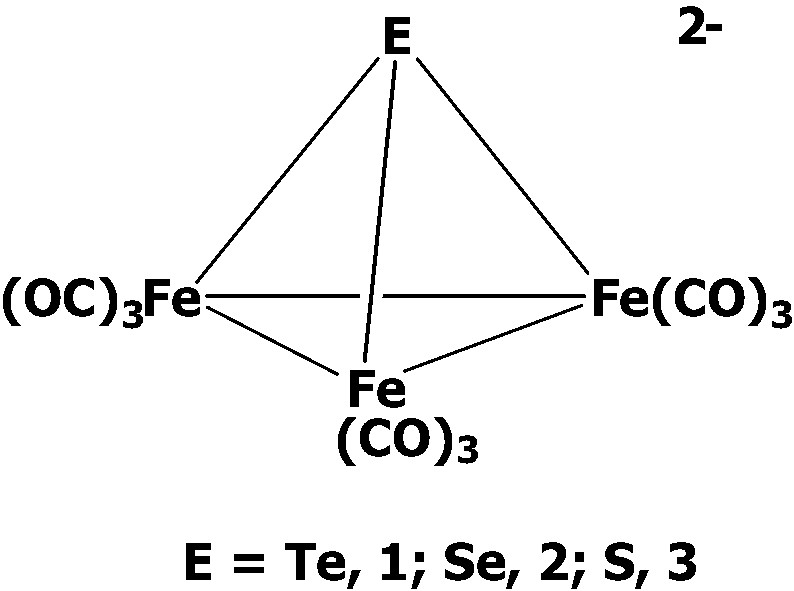
2 Synthesis of [EFe3(CO)9]2– (E = Te, Se, S) and the related cluster transformations
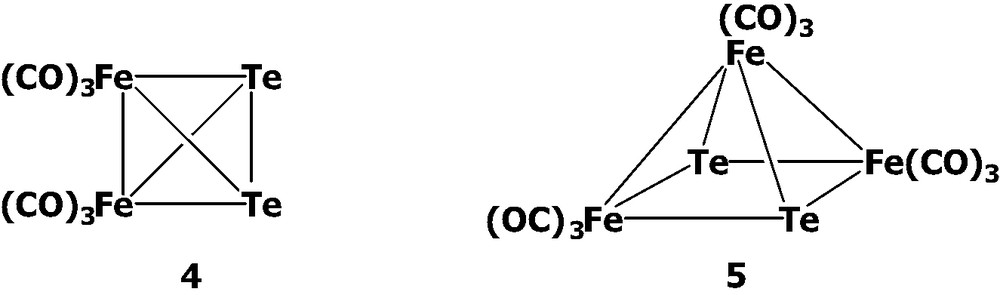
The syntheses of the chalcogen-containing triiron carbonyl clusters [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) were basically modified from the Hieber's synthesis [4]. Around 1950 s, Hieber et al. reported that TeO32– could react with iron carbonyl in basic solutions followed by acidification to produce the neutral diiron and triiron complexes Te2Fe2(CO)6 4 [5] and Te2Fe3(CO)9 5 [4a,6]. Since then, the reactivity patterns of these two complexes have been well studied [1,2]. However, before our study, there were no intermediate anionic clusters in this Te–Fe–CO system ever isolated because of their highly reactive natures. To control and understand better the cluster transformations in this Te–Fe system, we investigated the details of the Hieber's synthesis, and the extended study to the related Se and S systems has been carried out as well.
2.1 Synthesis of [TeFe3(CO)9]2– and Its cluster growth processes
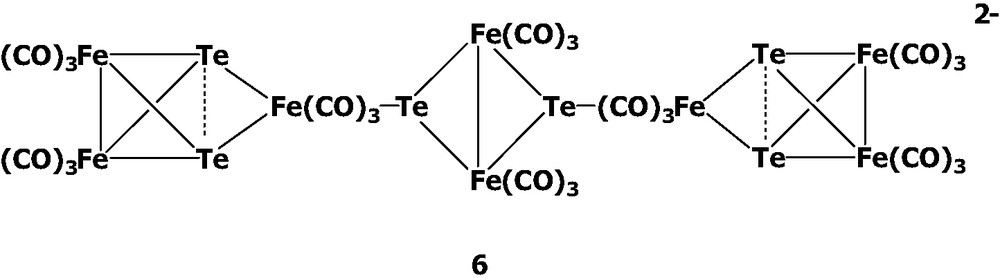
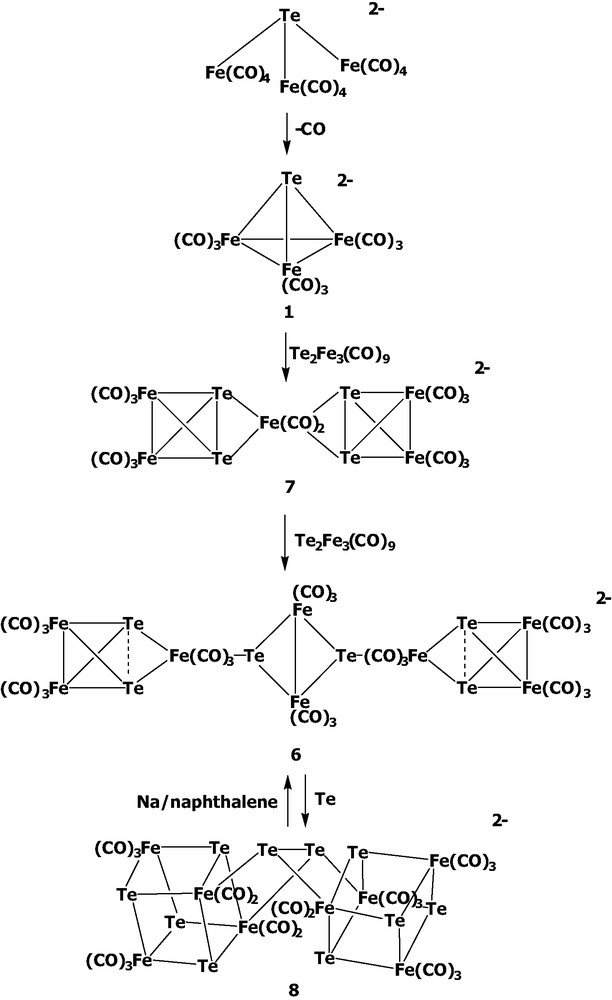
In the Te–Fe–CO system, the first anionic cluster [Te6Fe8(CO)24]2– 6 was isolated in our laboratory from the reaction of K2TeO3 with Fe(CO)5/KOH in MeOH solutions [7]. Cluster 6 exhibits the novel structural combination of the central diiron ditelluride and two terminal triiron ditelluride moieties. That is, cluster 6 can be viewed as two asymmetric Te2Fe3(CO)9 clusters attached to each of two Te atoms of the central anion [Te2Fe2(CO)6]2–.
Interestingly, it was found later that the different ratios of K2TeO3 with Fe(CO)5/KOH led to the formation of a series of anionic complexes [TeFe3(CO)12]2–, [TeFe3(CO)9]2– 1 [8], [Te4Fe5(CO)14]2– 7 [9,10], and [Te6Fe8(CO)24]2– 6. When the reaction of K2TeO3 and Fe(CO)5 was conducted in the ratio of 1:3, the triiron cluster [TeFe3(CO)12]2– was obtained, which displayed an open structure with all the carbonyls terminally coordinated. The open complex [TeFe3(CO)12]2– could then subsequently lose three carbonyls to form three Fe–Fe bonds giving the closo-triiron cluster [TeFe3(CO)9]2– 1 (Eq. 1). Surprisingly, as the ratio of Te to Fe was increased, the medium-sized cluster [Te4Fe5(CO)14]2– and the larger-sized cluster [Te6Fe8(CO)24]2– 6 were obtained. The isolation of these anionic clusters clearly indicated the molar ratio of K2TeO3 to Fe(CO)5/KOH is an important factor in determining which anionic species to be obtained, which suggested the cluster growth from the small triiron clusters to the larger-sized ones could be controlled.
To further understand and design the cluster growth processes, the following reactions were carried out. When cluster 1 was treated with Te2Fe3(CO)9, the medium-sized cluster [Te4Fe5(CO)14]2– 7 was obtained. One can envision [Te4Fe5(CO)14]2– 7 to be composed of the anion [Te2Fe2(CO)6]2– and one Te2Fe3(CO)9 with loss of one CO, which seems to have no direct structural relevance to [TeFe3(CO)9]2– and Te2Fe3(CO)9. Therefore, [Te4Fe5(CO)14]2– 7 was considered to result from complicated bond breakage and rearrangement processes. It was of interest to note that [Te4Fe5(CO)14]2– 7 could further react with Te2Fe3(CO)9 to yield the large cluster [Te6Fe8(CO)24]2– 6 (Eq. 2). This cluster growth process is quite reasonable, because cluster 6 also can be approximately viewed as one [Te4Fe5(CO)14]2– anion and one Te2Fe3(CO)9 molecule.
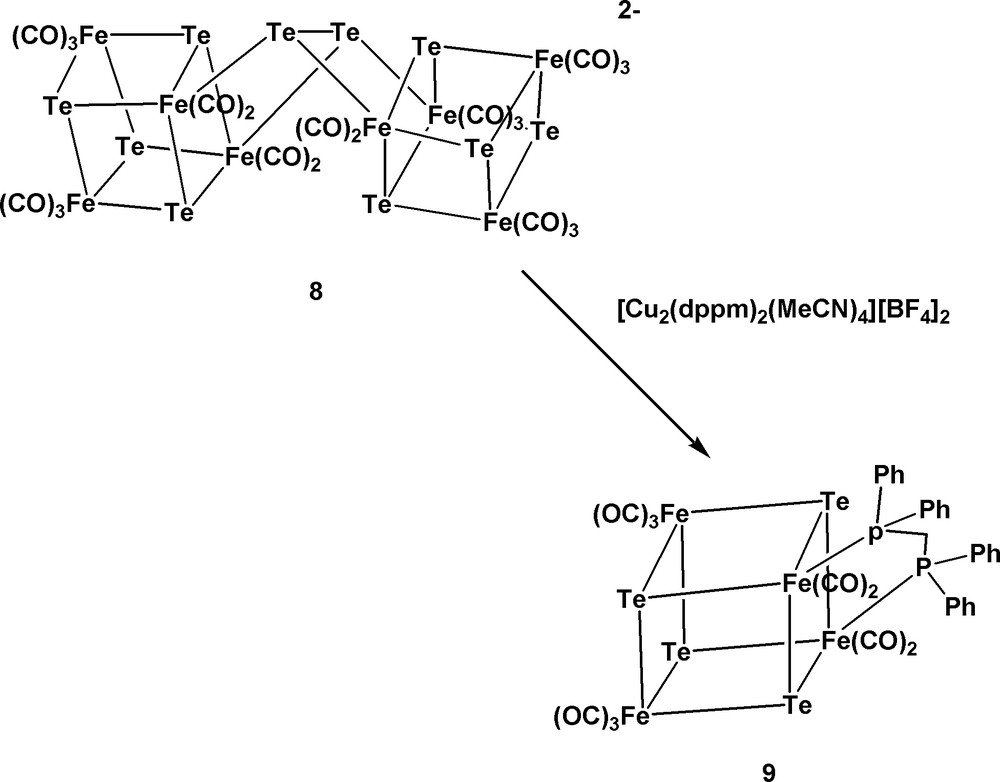
In addition, when 6 was further treated with 4 equiv of Te, the double-cubane cluster [Te10Fe8(CO)20]2– 8 was formed in good yield (Eq. 3) [11]. Cluster 8 was once reported to be generated from the reaction of Fe(CO)5 with the ‘Zintl’ ion Te42– and excess Te [9]. However, the route to cluster 8 from 6 is more convenient and gives higher yield. It was also found that cluster 8 can be reconverted to cluster 6 upon reduction with Na/naphthalene. From all these structural transformations, it is concluded that the cluster growth from complex 1 to the large-sized clusters 6 and 8 is feasible and can be easily controlled (Scheme 1). Other larger-sized clusters might be designed and synthesized along these lines.
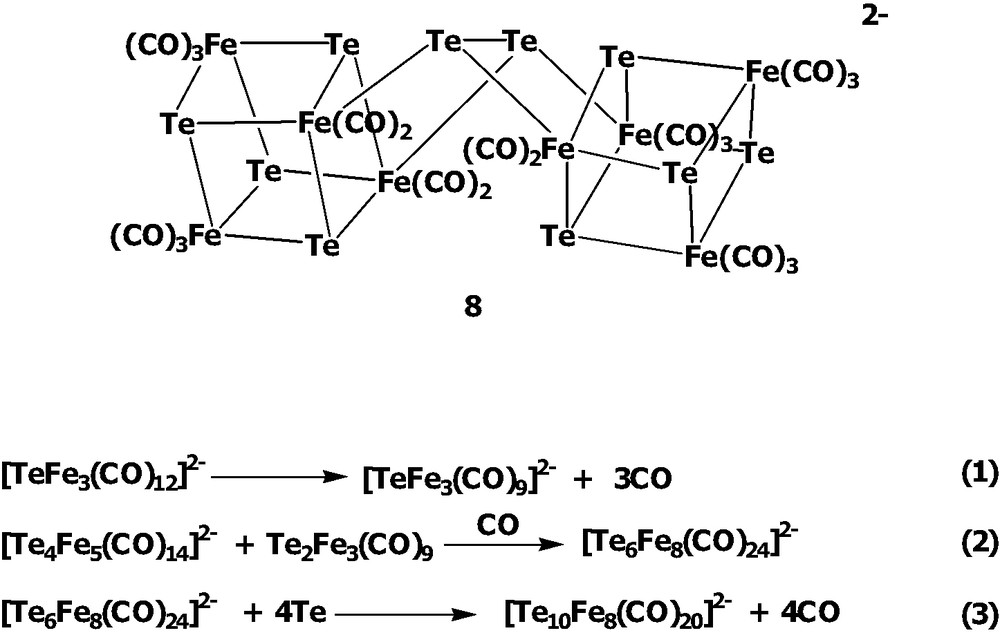
Subsequently, when [Te10Fe8(CO)20]2– 8 was treated with [Cu2(dppm)2(MeCN)4][BF4]2, the dppm-bridged cubic cluster [Te4Fe4(CO)10(dppm)] 9 was obtained (Scheme 2) [11]. Cluster 9 can be considered to result from the oxidative fragmentation of 8 by the Cu(I) complex followed by the chelation of the dppm ligand. On the other hand, the reaction of cluster 8 with MeI gave the double-methylated fragmented product Fe2(CO)6(TeMe)2 [12]. These results showed that the double cubic cluster 8 tended to undergo fragmentation to give the cubic or even smaller fragments depending upon the incoming reagents.
2.2 Synthesis of [SeFe3(CO)9]2– and Its cluster growth processes
Similar to what was observed in the Te–Fe system, the reaction of K2SeO3 with Fe(CO)5/KOH in various ratios produces a series of anionic species as the intermediates which can be further oxidized by the appropriate agents to form the tetrahedral cluster Se2Fe2(CO)6 10 [13] and the square pyramidal complex Se2Fe3(CO)9 11 [14].
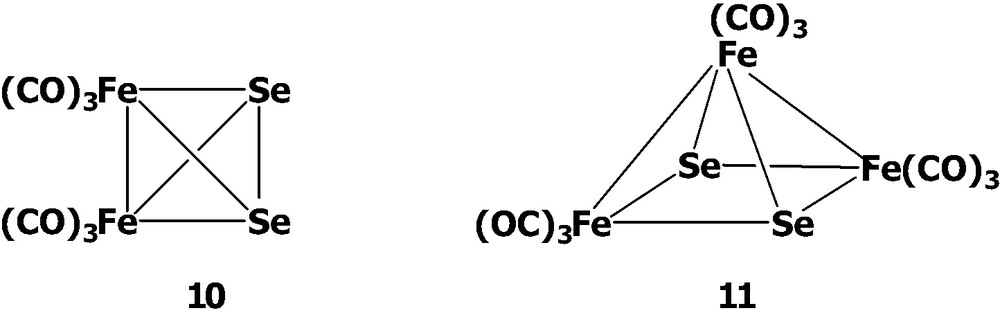
The interesting structural transformations of the anionic species in the Se–Fe–CO system have been established by the variation of the concentration of K2SeO3 in the Hieber's synthesis (Scheme 3). The reaction was carefully done by employment of K2SeO3 with Fe(CO)5/KOH in MeOH solution. It has been demonstrated that the open complex [SeFe3(CO)12]2– can transform to the tetrahedral cluster [SeFe3(CO)9]2– 2 by the loss of three carbonyls and to the reactive species [(Se2)2Fe2(CO)6]2–. After prolonged stirring, as Fe(CO)5 taking up a total of 1 equiv of K2SeO3, [SeFe3(CO)9]2– 2 can further couple with [(Se2)2Fe2(CO)6]2– to form the medium-size cluster [Se6Fe6(CO)12]2– 12 [15]. Cluster 12 can be viewed as two Se2Fe2(CO)6 clusters asymmetrically attached each to two Fe atoms of the central anion [Se2Fe2]2–. Cluster 12 decomposes to give 10 upon acidification and can be oxidized by [Cu(MeCN)4]BF4 to form cluster 11.
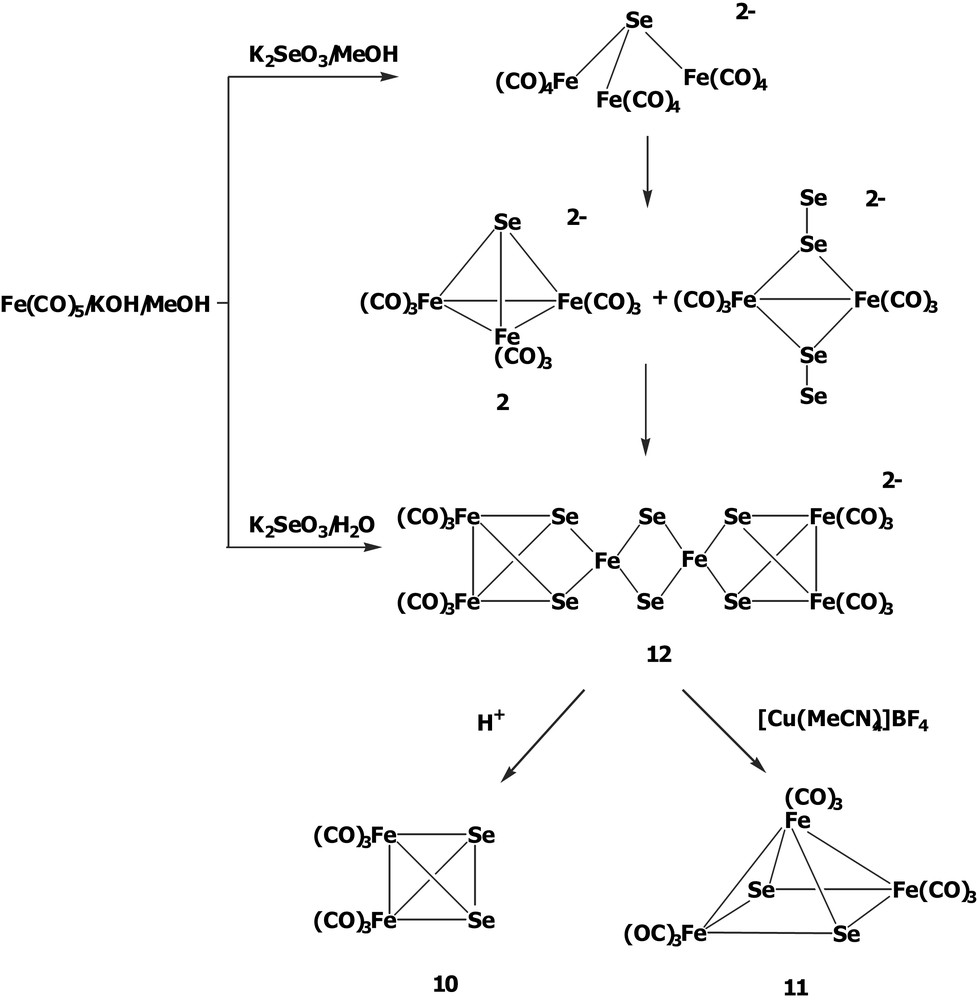

In this Se–Fe–CO system, the cluster growth processes from cluster 2 to cluster 12 are apparently dependent on the molar ratios of K2SeO3 to Fe(CO)5. Other larger-sized clusters might be expected by appropriately controlling the ratio of the reactants and the reaction conditions.
2.3 Synthesis of [SFe3(CO)9]2–
Before our study, the tetrahedral cluster anion [SFe3(CO)9]2– 3 has been reported from several routes; however, those preparations either result in complex mixtures of products or need special reaction conditions [16]. We developed a facile synthesis of [SFe3(CO)9]2– 3 by the treatment of Na2SO3 with Fe(CO)5/KOH in refluxing methanol solution. The reaction gives a moderate yield under milder conditions. If this reaction was not conducted in basic solution, the monohydrido cluster [HSFe3(CO)9]– 13 would be produced along with the major product [SFe3(CO)9]2– 3.
3 Reactivity of [EFe3(CO)9]2– (E = Te, Se, S) toward inorganic and organic fragments
In this section, we will describe the reactivities of [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) with a series of inorganic and organic fragments. The incoming species include Ru3(CO)12, some electrophiles (e.g. haloalkanes and inorganic salts), and the bifunctional propargyl bromide HC≡CCH2Br.
3.1 Reactions of [EFe3(CO)9]2– (E = Te, Se, S) with Ru3(CO)12
While the homonuclear transition metal clusters bridged by main group elements have been well developed, the heteronuclear metal clusters are comparatively less explored [1,2,17,18]. In order to explore the cluster expansions, the reactions of [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) with heteronuclear metal carbonyl Ru3(CO)12 were investigated in our laboratory.
It has been shown that [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) can react with Ru3(CO)12 in refluxing acetone to undergo skeletal expansion forming the mixed-metal octahedral clusters [(μ4-E)Fe2Ru3(CO)14]2– (E = Te, 14; Se, 15; S, 16) [19–21] (Scheme 4). Clusters 14–16 each display a Fe2Ru2 plane that is further capped above and below by one Ru(CO)3 group and one E ligand, respectively.
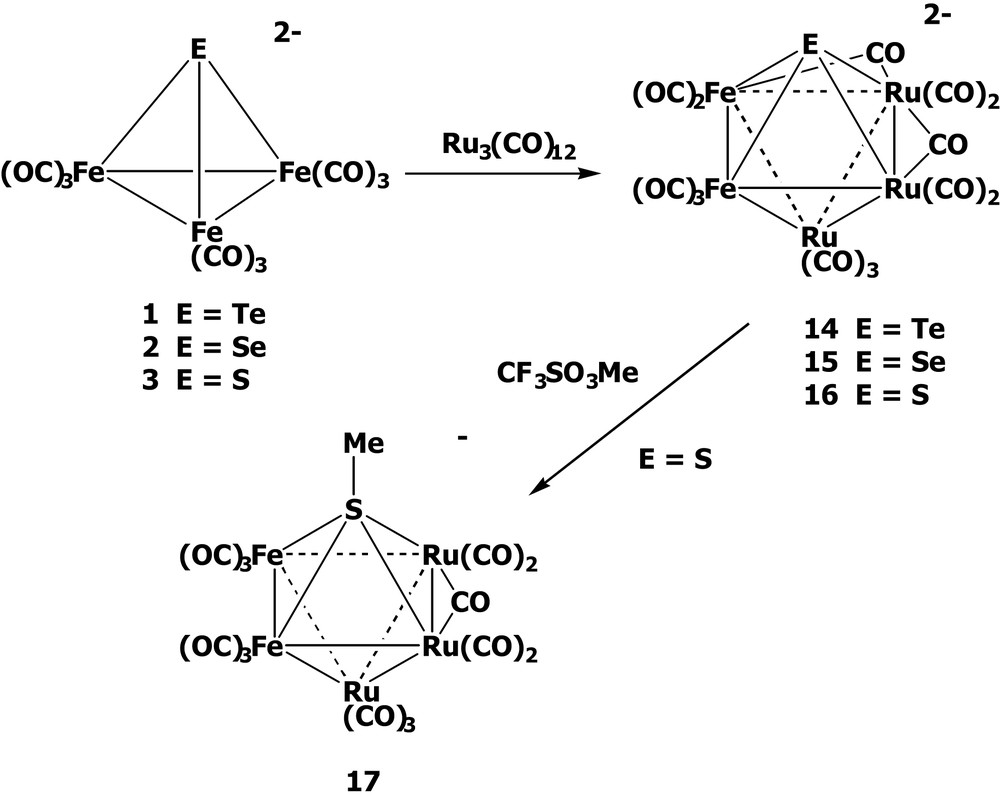
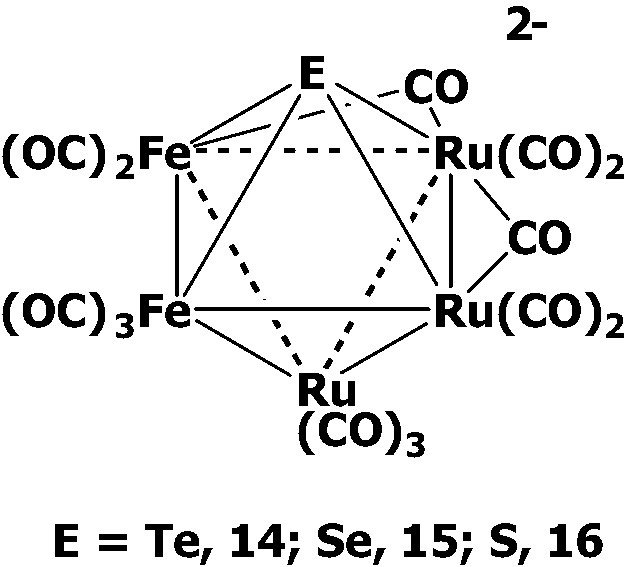
To further test the basicity of the sulfur atom in the cluster [(μ4-S)Fe2Ru3(CO)14]2– 16, we treated the octahedral cluster 16 with CF3SO3Me. It was found that the careful methylation led to the formation of the sulfur-methylated octahedral clusters [MeSFe2Ru3(CO)14]– 17 [21], in which the sulfur atom is pentacoordinated to one methyl group and two Ru and two Fe atoms, indicative of the basic character of the μ4-S atom (Scheme 4). However, the methylation of the octahedral clusters [(μ4-E)Fe2Ru3(CO)14]2– (E = Te, 14; Se, 15) failed to give the methylated products due to the weaker E–C bonds.
3.2 Reactions of [EFe3(CO)9]2– (E = Te, Se, S) with electrophiles
To systematically investigate the reactivity patterns toward electrophiles, the anionic clusters [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) were treated with a series of organic halides and inorganic salts. These reactions appear to be useful synthetic routes for the preparation of chalcogen-containing clusters incorporated with various combinations of organic fragments and/or transition metals.
3.2.1 [TeFe3(CO)9]2– with electrophiles
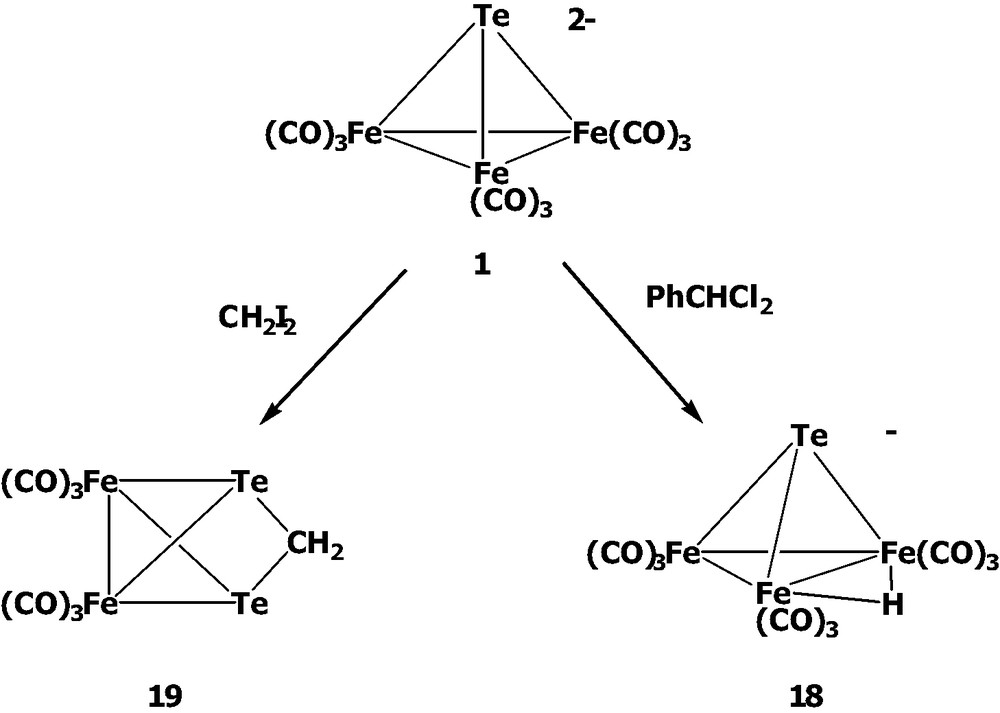
[TeFe3(CO)9]2– 1 has been reported to react with acids to form the Fe–Fe bridged monohydrido cluster [HTeFe3(CO)9]– 18 and the dihydrido cluster H2TeFe3(CO)9 [22,23]. Cluster 1 can also be easily protonated in MeOH to form the monohydrido cluster 18 [24]. Similar to the protonation, the treatment of 1 with PPh3Au+ or PhHg+ yields the Fe–Fe bridged clusters [TeFe3(μ-AuPPh4)]– and [TeFe3(μ-HgPh)]–, respectively [23]. On the other hand, the reaction of 1 with CuCl forms the Fe–Fe bridged cluster [TeFe3(CO)9(μ-CuCl)]2– with CuCl intact [22]. Upon the addition of NOBF4, cluster 1 was found to undergo the CO substitution with NO+ to give cluster [TeFe3(CO)8(NO)]–. When cluster 1 was further treated with I2, the complicated disproportionation reaction occurred to form the butterfly cluster [Te2Fe2(CO)6(μ-Fe(CO)3I)]– [23]. Recently, a number of novel nido-clusters of the type [Fe3(CO)9(μ3-Te)(μ3-ER)] containing Te and group 15 element (E = As, Sb) with attached functional groups (R = Me, 2,4,6-Me3C6H2, Cp*, (η5-C5H4tBu)Fe(CO)2) have been obtained by the reaction of cluster 1 with REX2 (X = Br, I) [25]. Cluster 1 could also undergo cluster expansion with [Cp*M(MeCN)3][CF3SO3]2 (M = Rh, Ir) to produce the mixed-metal products [MFe3(μ4-Te)(CO)9Cp*] (M = Rh, Ir) and [IrFe2(μ3-Te)(CO)7Cp*], respectively [26].
The reactions of [TeFe3(CO)9]2– 1 with some other organic dihalides were also performed in our laboratory (Scheme 5) [19]. When 1 was treated with CH2I2, the methylene-bridged cluster Fe2(CO)6(μ-TeCH2Te) 19 was obtained as a result of methylenation across the Fe–Fe bond. Instead of yielding the Fe-Fe bridged product, the similar reaction with PhCHCl2 produced the oxidized product [HTeFe3(CO)9]– 18 probably due to the larger size of the PhCH group vs. the CH2 group. The results suggested that the reactivity patterns of 1 towards electrophiles greatly depend on the size and the electrophilicity of incoming groups.
3.2.2 [SeFe3(CO)9]2– with electrophiles
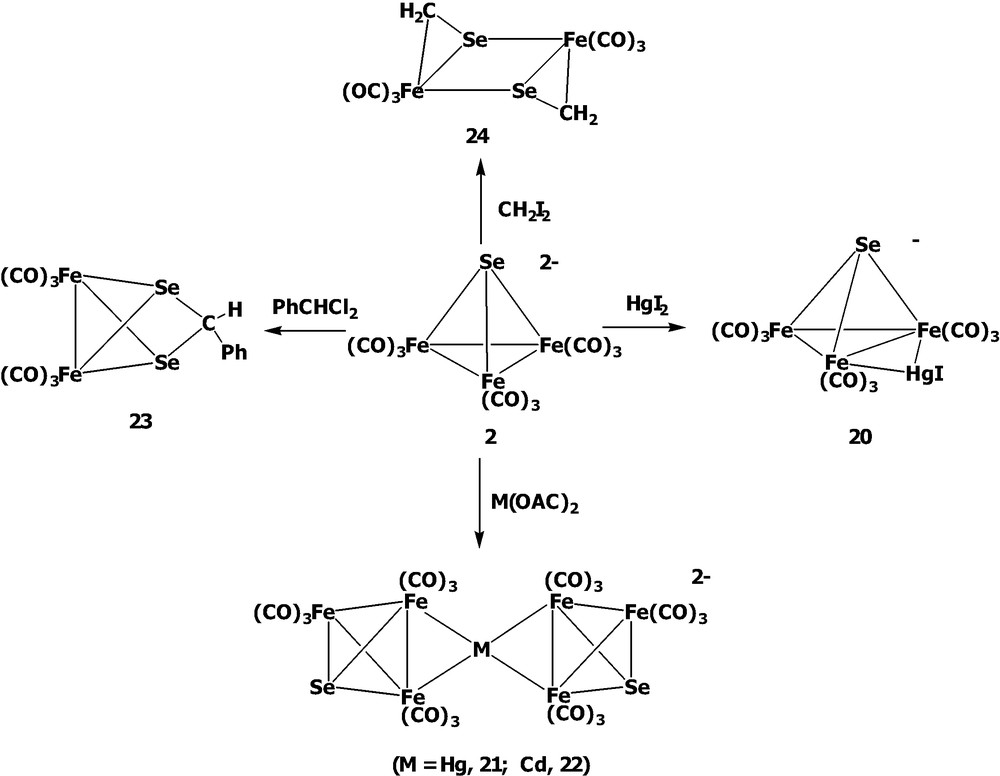
To compare with the tellurium system, the reactivities of the analogous [SeFe3(CO)9]2– 2 towards a series of electrophiles were also extensively studied in our laboratory (Scheme 6). Similar to the tellurium system, cluster 2 can also be protonated across the Fe–Fe bond to form the monohydrido complex [Se(μ-H)Fe3(CO)9]– and the dihydrido product Se(μ-H)2Fe3(CO)9 [21]. The monohydride complex [Se(μ-H)Fe3(CO)9]– was also known to be obtained when 2 was dissolved in methanol [24]. Further, when [SeFe3(CO)9]2– 2 was treated with HgI2, the HgI-bridged cluster [SeFe3(CO)9(μ-HgI)]– 20 could be produced. In contrast, the μ4-M clusters [{SeFe3(CO)9}2M]2– (M = Hg, 21; Cd, 22) were formed as complex 2 reacted with Hg(OAc)2 or Cd(OAc)2 [26] due to the different degree of ionization of the incoming transition metal salts. On the other hand, while the reaction with CHPhCl2 produced the Se–Se bridged butterfly cluster Fe2(CO)6(μ-SeCHPhSe) 23, the treatment with CH2I2 formed the Se–Fe bridged ring product Se2Fe2(CO)6(μ-CH2)2 24. The differing reactivities toward these organic dihalides could be attributable to the size effect of the incoming organic groups [20].
Recently, it has been reported that the triiron clusters [Fe3(CO)9(μ3-Se)(μ3-ER)] and Fe3(CO)10(μ3-η2:η1-SeSbMes) incorporated with mixed Se–group-15 element (E = As or Sb) bonded with the functional groups (R = Me, 2,4,6-Me3C6H2, Cp*, or (η5-C5H4tBu)Fe(CO)2) could be obtained by the reaction of 2 with REX2 (X = Br, I) [25]. Furthermore, the triiron-based complex [Fe3(μ3-BiFe(CO)2(η5-C5H4tBu))(μ3-Se)(CO)9] containing a Bi-Se combination has also been synthesized in a similar manner [25c]. Cluster 2 can also undergo cluster expansions with [Cp*M(MeCN)3][CF3SO3]2 (M = Rh, Ir) to give the mixed-metal neutral products [MFe3(μ4-Se)(CO)9Cp*] (M = Rh, Ir) and [IrFe2(μ3-Se)(CO)7Cp*] [26], respectively.
3.2.3 [SFe3(CO)9]2– with electrophiles
The tetrahedral cluster [SFe3(CO)9]2– 3 is isostructural with its analogs [EFe3(CO)9]2– (E = Te, 1; Se, 2; O) [22–24,27,28]. Similar to the Te and Se cases, most reactions of complex 3 with electrophiles were reported to occur across the Fe–Fe bond, and such examples included [Fe3(μ3-S)(μ-AuPPh3)(CO)9]– [29] (PPh3Au)2Fe3(CO)9S [30,31], and Fe3S(X)(CO)9 (X = μ2-SnMe2, μ3-PPh) [31].
As mentioned above, we developed a new synthetic route to [SFe3(CO)9]2– 3 by the employment of Na2SO3 with Fe(CO)5/KOH in refluxing methanol solution, and its subsequent reactions with some electrophiles were also investigated (Scheme 7) [21]. Following the similar reactivity pattern, complex 3 could be protonated across the Fe–Fe bond to form the monohydrido cluster [SFe3(μ-H)(CO)9]– 13 and the dihydrido cluster SFe3(μ-H)2(CO)9 25. In contrast, complex 3 could react with CF3SO3Me to give the sulfur-methylated cluster [MeSFe3(CO)9]– 26, indicative of the greater affinity of the sulfur atom vs. the iron centers toward the methyl group. Similarly, 3 was reported to react with the cationic complexes [Mn(CO)3(MeCN)3]+ or [Re(CO)5]+ to form the μ4-sulfido clusters [Fe3(CO)9(μ4-S)M(CO)5]– (M = Mn, Re) [17]. Further, the sulfur atom of 3 was found to be alkylated by some alkyl halides followed by acidification giving complexes HFe3(CO)9(SR) [32]. Instead of forming the methylenation product, the reaction of 3 with CH2I2 led to the formation of the oxidized product [SFe3(μ-H)(CO)9]– 13. As in the Te and Se systems, complex 3 could be dissolved in methanol to form [HSFe3(CO)9]– 13 [24].
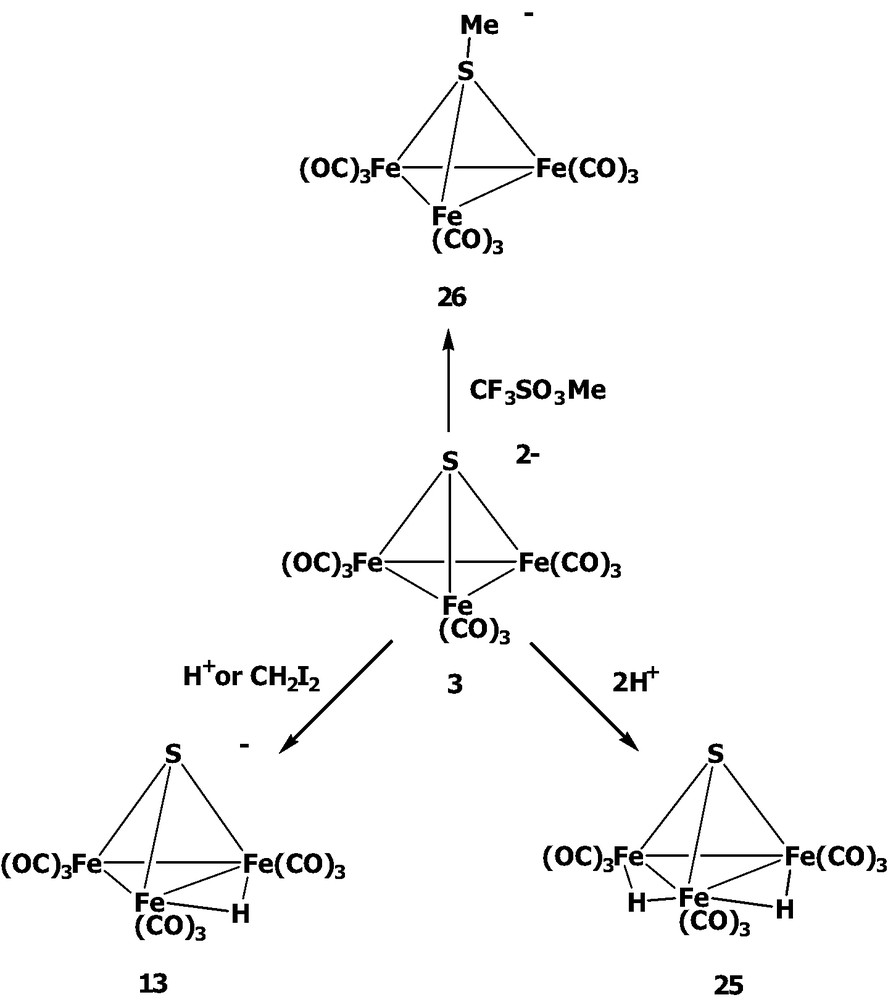
Based on the versatile outcomes of the reactions of [EFe3(CO)9]2– (E = Te, 1; Se, 2; S, 3) with such a wide range of electrophiles, it is summarized that the complicated coupling effects of chalcogen and incoming electrophiles should play the determining role in the reactive sites and resultant clusters in these reactions in terms of the electronic and steric considerations.
3.3 Reactions of [EFe3(CO)9]2– (E = Te, Se) with the bifunctional propargyl bromide
While main group-transition metal carbonyl clusters have attracted attention, their interaction with organic moieties was rarely explored [1,2]. We have explored the reactions of [EFe(CO)9]2– (E = Te, 1; Se, 2; S, 3) with the bifunctional propargyl bromide to probe the possible reactive sites in many industrially important metal-surface catalyzed reactions [24,33]. In our study, the reactions with propargyl bromide are generally more complicated than those with alkyl halides and usually require careful treatments. Our studies showed that the similar reaction patterns with propargyl bromide could be observed in the Se and Te systems but not for the S system probably owing to the similar metallic property of Te and Se. However, other complex coupling factors could also play the key role in controlling the results. In this section, we will describe the reactions of clusters 1 and 2 with propargyl bromide [34,35], and the reactions in the S system will be discussed elsewhere.
3.3.1 [SeFe3(CO)9]2– with propargyl bromide
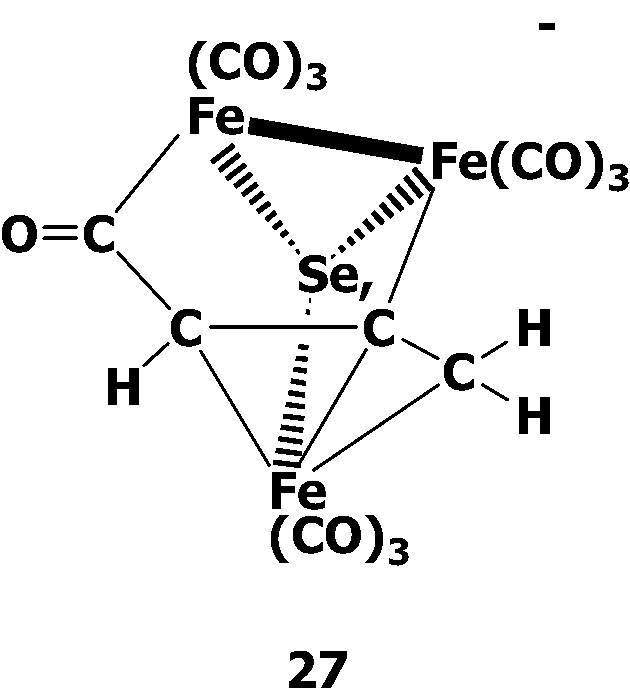
When [SeFe3(CO)9]2– 2 was treated with 1 equiv of propargyl bromide HC≡CCH2Br in MeCN, the novel acyl complex [(μ3-Se)Fe3(CO)9(μ3-η1:η1:η3-C(O)C(H)CCH2)]– 27 was obtained. The anion 27 was found to consists of three Fe centers with one Fe–Fe bond, in which three Fe atoms are capped on the opposite sides by a Se atom and by an allyl carbonyl ligand C(O)CHCCH2 in the fashion of μ3-η1:η1:η3. Cluster 27 can be considered as a CO insertion product, which is supported by the fact that the yield of 27 is significantly increased (54–85%) when the reaction is performed under an atmosphere of CO. Since 27 is an acylate complex, the rational synthesis of a Fischer-type iron cluster carbene by O-alkylation, was therefore, attempted. It was found that the reaction could succeed with CF3SO3Me to form two isomeric cluster carbene complexes trans-(μ3-Se)Fe3(CO)9(μ3-η1:η1:η3-C(OMe)CHCCH2) 28 and cis-(μ3-Se)Fe3(CO)9(μ3-η1:η1:η3-C(OMe)CHCCH2) 29 (Scheme 8). Surprisingly, complexes 28 and 29 could be separated by chromatography and were fully characterized by spectroscopic methods and single-crystal X-ray analysis.
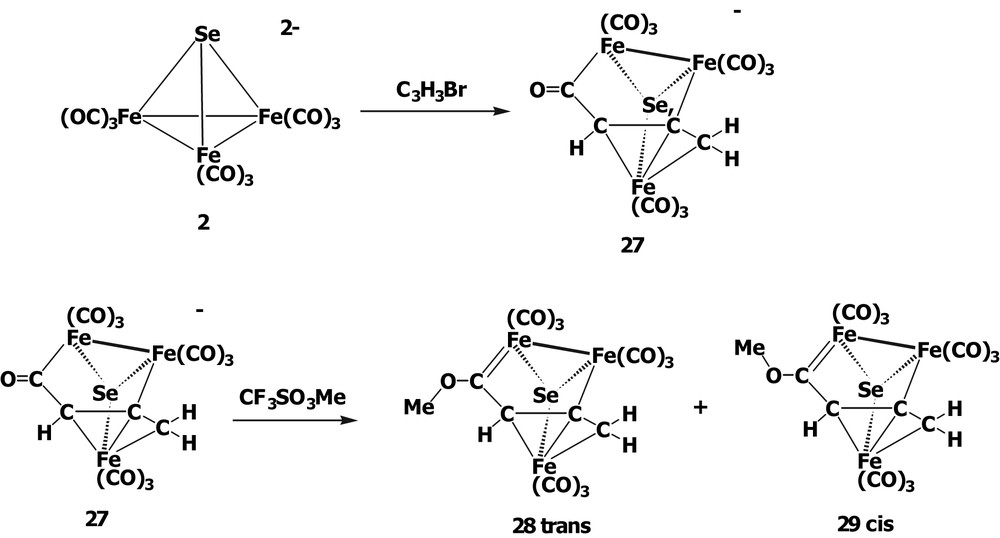
Complexes 28 and 29 can undergo cis/trans isomerization at room temperature. The line shape analysis of the VT NMR spectra gave the rotational energy of this cis/trans conversion, approximately 22.6 kcal/mol (94.6 kJ/mol), which is greater than those (11.5–13.8 kcal/mol) for the chromium methoxycarbene complexes [36] and permits the isolation of 28 and 29 at room temperature. Such an interesting phenomenon is unprecedented in the mononuclear Fischer-type alkoxycarbene complexes. This study reveals that the presence of the Se–Fe–CO cluster moiety and the extended organic network have a crucial influence on the rotation of the C–O bond of the methoxycarbene and allow for the successful isolation of the rotational isomers.
3.3.2 [TeFe3(CO)9]2– with propargyl bromide
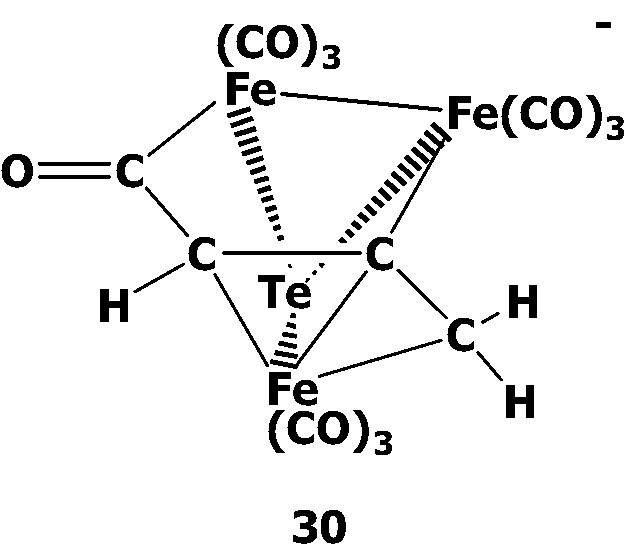
The varied ratios of [TeFe3(CO)9]2– 1 with HC≡CCH2Br were investigated in MeCN or CH2Cl2 (Scheme 9). When cluster 1 was treated with 1 equiv of propargyl bromide in MeCN, the novel acyl complex [(μ3-Te)Fe3(CO)9(μ3-η1:η1:η3-C(O)C(H)CCH2)]– 30 was obtained. Cluster 30 is isostructural with 27 and can be viewed to consist of a TeFe3 core linked by a novel allyl carbonyl ligand C(O)CHCCH2 in the fashion of μ3-η1:η1:η3. When 1 was treated with excess propargyl bromide in MeCN, two new butterfly clusters Te2Fe2(CO)6(CH=C=CH2)2 31 and Te2Fe2(CO)6(CH2C≡CH)(CH=C=CH2) 32 were obtained along with cluster 30. While clusters 31 displays a Te2Fe2 metal core with two allenyl ligands attached to the tellurium atoms in the anti position, cluster 32 consists of a Te2Fe2 core with each tellurium atom coordinated with one allenyl or one propargyl ligand in the anti position. Further investigation of cluster 1 with propargyl bromide in the molar ratio of 1: 2 or 1: 4 in CH2Cl2 showed that an oxidation product (μ3-Te)Fe3(CO)9(μ3-η1:η2:η1-C(Me)=CH) 33 was afforded in addition to the formation of 30, 31, and 32. Cluster 33 can be viewed to be composed of a TeFe3 core coordinated with a propyne ligand C(Me)≡CH which acts as a 4e– donor.
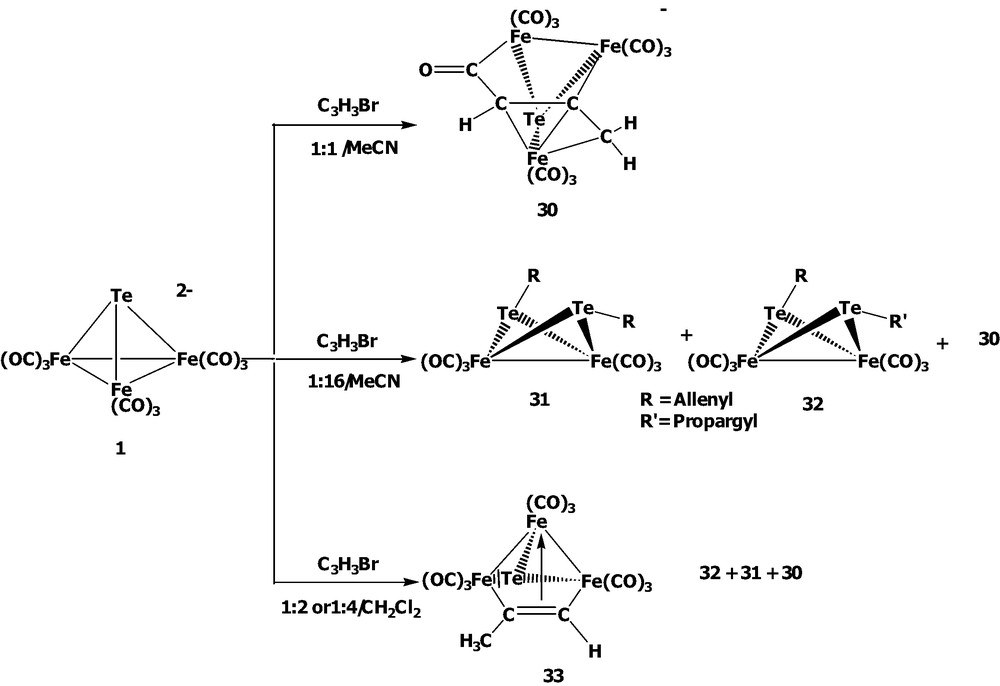
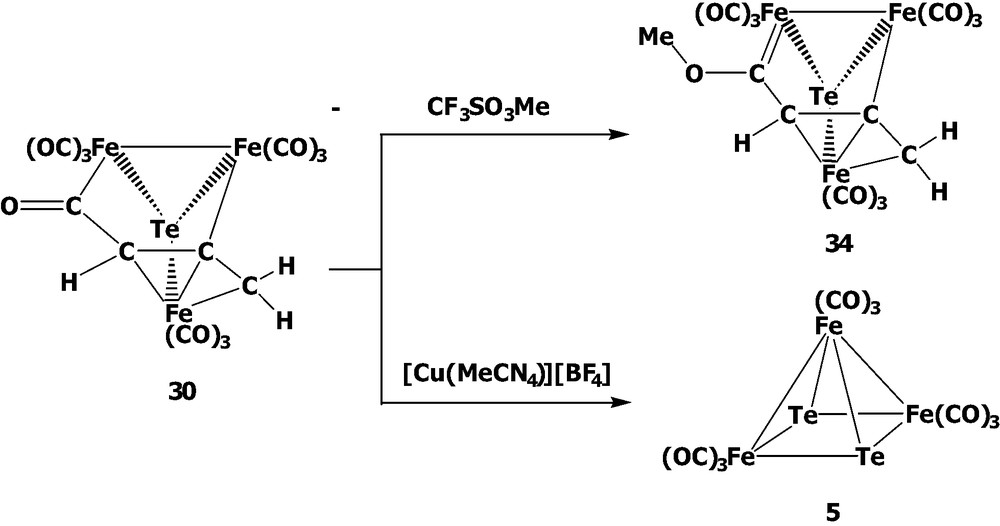
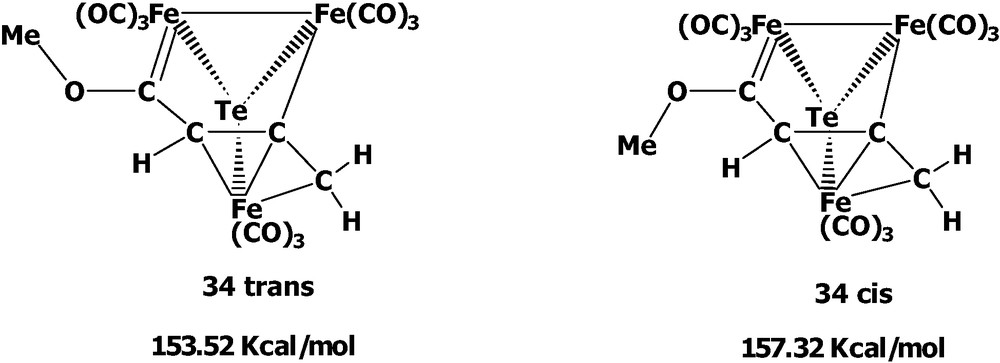
The reactivity of the acyl-cluster 30 was also investigated (Scheme 10). When cluster 30 was treated with [Cu(MeCN)4]BF4, the cluster fragmentation occurred via the organo ligand loss and the bond rearrangement to give the oxidation product Te2Fe3(CO)9 5. Interestingly, when cluster 30 was methylated with CF3SO3Me, the Fischer-type carbene complex (μ3-Te)Fe3(CO)9(μ3-η1:η1:η3-C(OMe)C(H)CCH2) 34 was afforded. Cluster 34 is isolated as a trans form in the solid state but the cis product is not obtained under our conditions due to the greater stability of the trans form, in which the geometric and energy minimization calculations are supported by the INSIGHT II (Discover) program (Scheme 11). This is contrary to the related Se system in that the cis and trans forms are both isolable at room temperature due to the greater rotational energy. These results suggest that the tellurium Fischer-carbene complex should have the higher degree of free rotation about the O–Ccarbene bond compared with the corresponding selenium complex. This phenomenon could be rationalized by the better metallic character of the tellurium atom vs. the selenium atom, which induces the less electron demand in the Fe–Ccarbene bond than in the Se carbene case.
4 Conclusion
In our previous investigations, we have not only developed the facile routes to the chalcogen-containing triiron carbonyl clusters [EFe3(CO)9]2– (E = Te, Se, S) but also studied their versatile reactivities towards a wide range of organic and inorganic electrophiles. More importantly, some intriguing quintessence has been concluded from this study. First, the stepwise cluster growth processes can be carefully controlled, which could provide some accessible routes to the nano-sized metal clusters. Secondly, the reactions of [EFe3(CO)9]2– (E = Te, Se, S) with appropriate electrophiles provide valuable routes to a series of homonuclear or heteronuclear clusters which may contain various combinations of organic, inorganic moieties, or mixed transition metal fragments. Further, the coupling effects of chalcogen and incoming electrophiles play the determining role in the outcomes of the nucleophilic reactions of [EFe3(CO)9]2– (E = Te, Se, S). In other words, the affinity of the chalcogen or iron centers towards the electrophiles may be logically determined on the basis of hardness/softness of the incoming groups.
Acknowledgements
The writing of this review was supported by National Science Council of Taiwan (NSC 90-2113-M-003-018 to M. Shieh) and by National Taiwan Normal University (ORD92-2).