

1 Introduction
There is considerable interest in heterobimetallic complexes containing lanthanide and transition metals. They have potential uses as new catalysts [1] or precursors for new materials, such as electroceramic and chemical sensors [2], fluorescent, magnetic properties [3] and rare earth orthoferrite materials [4].
Over the last few years, this laboratory has systematically synthesized and characterized a variety of lanthanide–transition metal (Ln–M) cyanide complexes [5]. These include new types of extended networks that have various dimensionalities, including chains, layers, columns, and anionic arrays with lanthanide cations sequestered in pockets. While principles of syntheses have been discovered and applied in the reproducible formation of new lanthanide–transition metal complexes, we have also recognized conditions that do not favor complex formation: a) a Ln–M direct metal–metal bond is apparently not formed due to the strong coordinating ability of the cyanide ligand to act as a bridge between metals, Ln–NC–M, b) no lanthanide cation and complex transition metal cyanide anion ion pairs have been isolated. Instead, cyanide bridges between the lanthanide and transition metals form. While the bridging connections between lanthanide and group 10 transition metals in general do not show any isomerism of the CN linkage, isomerism does occur in the linkage between group 11 transition metal Cu(I) atoms.
In this laboratory, in addition to systemizing syntheses and characterizing a wide variety of lanthanide–transition metal complexes, we converted precursor complexes to heterogeneous catalysts on oxide supports. The heterogeneous catalysts derived from Ln–M cyanide compounds have demonstrated improved activity and selectivity over transition metal only catalysts in some important catalytic processes, such as the reduction of nitrogen oxides [1a], vapor-phase hydrogenation of phenol [1b], and hydrodechlorination of chlorobenzenes [1c,d]. The polymeric structural framework of these Ln–M cyanide catalyst precursors enables the uniform dispersion of the metals over the support's surface [3a] which results in significant improvement in catalytic activities and stabilities. For this reason, the design of catalyst precursors with Ln–M extended array like structures has been a priority in this laboratory.
Relatively recently we have employed the carbonyl ligand in lanthanide–transition metal complexes. Advantages of this ligand are that it is neutral and softer than cyanide. It has been shown that the nucleophilicity [6] of a transition metal carbonylate anion can vary remarkably as a function of the transition metal. Furthermore, the carbonyl ligands also possess nucleophilic character in their ability to form isocarbonyl linkages with lanthanide metals. These transition metal carbonylate anions offer more possibilities than cyanide ligands for interactions between transition metal carbonylates and lanthanides. Finally, the basicity of solvents can direct the formation of the products.
Numerous lanthanide–transition bimetallic carbonyl complexes have been prepared and characterized. Until now, the determination of the structural relationships between the two metals has revealed three different Ln–M complexes (Chart 1): (a) solvent-separated ion pairs (I), (b) isocarbonyl linkages (II), and (c) Ln–M direct bonds (III).
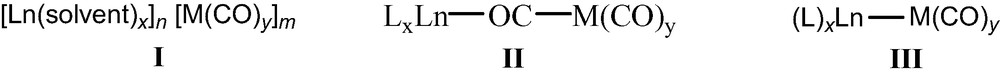
This paper will focus on lanthanide–transition metal carbonyl arrays.
2 Ln(II)–M carbonyls
Over the last two decades, studies of lanthanide(III)–transition metal carbonyl complexes have resulted in syntheses of the three types of interactions cited above [7]. These complexes contain anionic ligands on the lanthanide cation, LnLx(x = 2 usually, L = Cp–, etc.), that partially neutralize the charge of the cation which interacts with transition metal carbonylate anions to form stable complexes. All of the reported complexes have anionic ligands coordinated to Ln3+.
In this laboratory we are interested in complexes that only have solvent molecules coordinated to the lanthanide cation which interact with the transition metal carbonylate anion for potential applications in catalysis and materials chemistry. While anionic ligand-free Ln(III) cations do not tend to form stable Ln(III)–transition metal carbonyl complexes, divalent lanthanide cations which are softer Lewis acids, are more likely to form bimetallic complexes with transition metal carbonylates. In a similar vein, soft Lewis basic and strongly nucleophilic transition metal carbonylate anions are beneficial for a direct Ln(II)–M bond. The relative nucleophilicities of the metals of several [M(CO)y]n– anions were measured electrochemically by King et al.; the order decreases as: [CpFe(CO)2]– > [CpRu(CO)2]– > [CpNi(CO)]– > [Re(CO)5]– > [CpW(CO)3]– > [Mn(CO)5]– > [CpMo(CO)3]– > [CpCr(CO)3]– > [Co(CO)4]– [6]. [Fe(CO)4] [2–] has strong donating properties and was once dubbed a “supernucleophile” [8]. Even though there is no direct rate comparison [Fe(CO)4] [2–], is not any weaker than [CpFe(CO)2]– [9]. We find that the degree of interaction between Ln and M can be controlled by changing [M(CO)y]n– and solvent. For instance, an Ln–M bond is expected when the ‘supernucleophile’ [Fe(CO)4]2–, interacts with a lanthanide cation. Thus complexes that contain Yb–Fe bonds have been formed [10,11]. A solvent-separated ion pair ensues when the electron donating ability of the solvent to the lanthanide cation is stronger than the electron donating ability of the metal in [M(CO)y]–. An isocarbonyl linkage, Ln–OC–M, is afforded when the carbonyl oxygen is more nucleophilic than the M center. The result is the donation of an electron pair from a carbonyl oxygen to Ln.
2.1 Preparation of Ln–M carbonyls
Synthetic procedures toward Ln(II, III)–M carbonyl compounds were developed and refined, and the structural relationship of the metal combinations have been probed over the last three decades. Preparative methods for these heterometallics generally utilize simple adduct formation [12], metathesis reactions [7h,12c,13], transmetalation reactions (metal exchange) [7j,13b,14]. M–M bond cleavage (1-e– transfer [7a,b,d,e,f,k,13c], reduction in liquid ammonia [7m,10,11], amalgam reduction [15]), and M–X bond cleavage [16].
This laboratory has utilized most of the synthetic procedures indicated above to prepare Ln–M carbonyls. Of the lanthanide–transition metal carbonyls synthesized, ionic interactions, direct lanthanide–transition metal bonds and isocarbonyl linkages between Ln and M centers have been observed with structural types consisting of discrete molecules, one-dimensional, two-dimensional and polymeric arrays.
2.2 Solvent-separated ion pairs
When the weak nucleophiles [Co(CO)4]–, and [Mn(CO)5]– are engaged in reactions with lanthanide cations in the presence of strong coordinating solvents, the strong nucleophilicity of the solvent hinders the penetration of weak nucleophiles such as [Co(CO)4]– or [Mn(CO)5]– into the Ln2+ coordination sphere. Solvent-separated ion pair complexes are obtained [14].
Pyridine and THF are both stronger Lewis bases than [Co(CO)4]–. The transmetalation reaction of Yb with Hg[Co(CO)4]2 (Eq. (1)) in these solvents generates ion pairs, [Yb(L)6] [Co(CO)4]2 (1 and 2). During this reaction, Yb metal is oxidized to Yb(II) with concomitant reduction of Hg(II) to elemental Hg.
Lanthanide/mercury amalgam reduction of Co2(CO)8 or Mn2(CO)10 in DME (glyme) or DIME (diglyme) results in cleavage of the M–M bond in the carbonyl dimer and produces an Ln2+ cation and the [Co(CO)4]– or [Mn(CO)5]– anions (Eqs. (2) and (3)), with the production of similar ion pair complexes [Ln(L)x] [Co(CO)4]2 (3–5) and [Ln(L)x] [Mn(CO)5]2 (6–9). [14,15a] Fig. 1 shows the coordination geometry of the cation, [Sm(DME)3]2+, in complex 4.
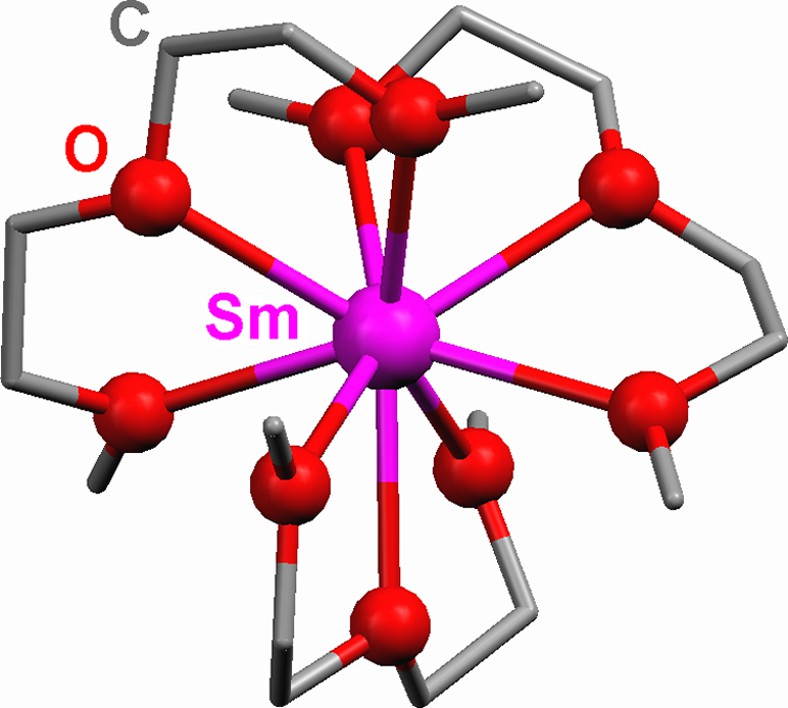
Molecular structure of the cation in [Sm(DIME)3] [Co(CO)4]2 (4). DIME ligands are drawn as capped sticks. H atoms are omitted for clarity.
On the other hand, a similar Yb/Hg amalgam reduction of Fe(CO)5 in CH3CN, generates an unexpected solvent-separated ion pair, [Yb(CH3CN)x] [Hg{Fe(CO)4}2] (10) [15a]. Compound 10 could not be crystallized in CH3CN. But [(DIME)2Yb(CH3CN)2] [Hg{Fe(CO)4}2] (11) and [(Pyr)5Yb(CH3CN)2] [Hg{Fe(CO)4}2]·2Pyr (12) were isolated in mixed solvents. During the reaction, [Fe(CO)4] [2–] and [Yb(CH3CN)x]2+ are generated initially, but the Hg prevents the formation of a Yb–Fe bond because [Fe(CO)4] [2–], a soft Lewis base, prefers the softer Lewis acid, Hg(II), over Yb(II).
Carbonyl oxygens may compete with solvent for binding to the lanthanide cation in solution. When the relative nucleophilicities of the carbonylate anion and solvent are comparable, this competition is expected, and an equilibrium between the ionic compound and an isocarbonyl complex arises. The presence of an isocarbonyl interaction is observable from solution IR spectroscopy. For instance, in THF, isocarbonyl associations are observed for 2, and the strength of the interaction is a function of concentration [14]. Dilute solutions of 2 have primarily the solvent-separated ion (1887 cm–1), but a weak contact ion pair ([(THF)xYb···OCCo(CO)3] [Co(CO)4]; 1898 and 1854 cm–1), also exists. More concentrated solutions show the emergence of a broad low frequency band at 1789 cm–1 and several higher frequency terminal CO stretches. Therefore, higher concentrations of 2 promote the formation of a strong carbonyl-bridged complex that is in equilibrium with the solvent-separated and weak contact ion pairs.
2.3 Isocarbonyls
As indicated in the preceding section, competing factors are involved in determining the formation of isocarbonyl or ion paired complexes. If the carbonyl oxygens are more Lewis basic than the transition metal center and the basicity of the anion is stronger than that of the solvent, then an isocarbonyl bridge between Ln and M is formed [7a,b,d,j,e]. Low polarity or non-nucleophilic solvents can also favor the formation of an isocarbonyl linkage even when [M(CO)y]n– is a weak nucleophile [7a,14].
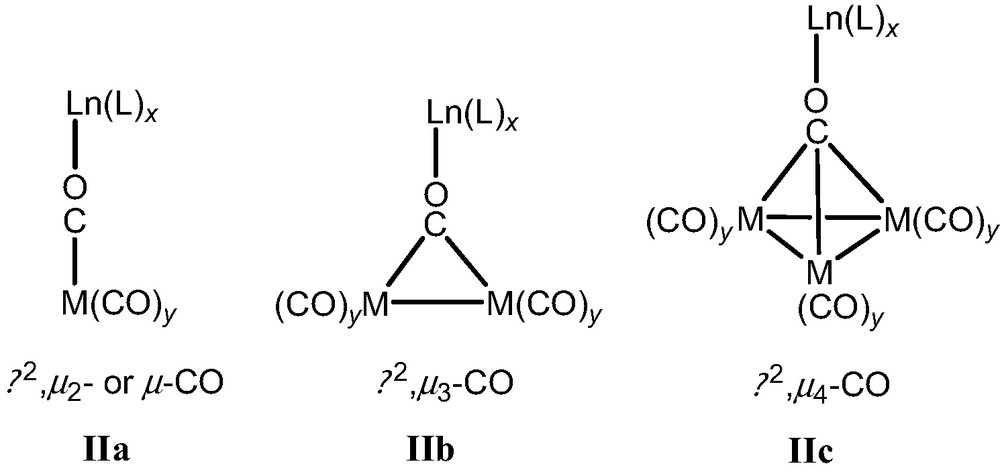
Two preparative pathways to isocarbonyl complexes have been applied in this laboratory: (a) direct reaction of lanthanide metal and Hg[(Co(CO)4]2 in a weakly nucleophilic solvent, (b) dissolving an ion-paired complex in a weakly nucleophilic solvent. Three types of isocarbonyl interactions can be expected based upon the types of carbonyls (Chart 2) of which two types of isocarbonyl interactions η2,μ2-CO (μ-CO; IIa) [7a,d,j,k,l,14] and η2,μ3-CO [7b,e] (IIb) are known. Complexes with η2,μ4-CO bridges (IIc) have only recently been reported [17].
2.3.1 Direct method and the formation of cobalt carbonyl clusters
Transmetalation reactions (Eq. (4)) involving Ln metal (Ln = Yb, Eu) and Hg[Co(CO)4]2 were carried out in diethylether. This solvent is sufficiently weakly basic that isocarbonyl linkages formed in the complexes {(Et2O)3Ln[Co4(CO)11]}∞ (13, Ln = Yb; 14, Ln = Eu; [(Et2O)3Ln{(η2,μ4-CO)(η2,μ3-CO)2Co4(μ2-CO)(CO)7}]∞) [17].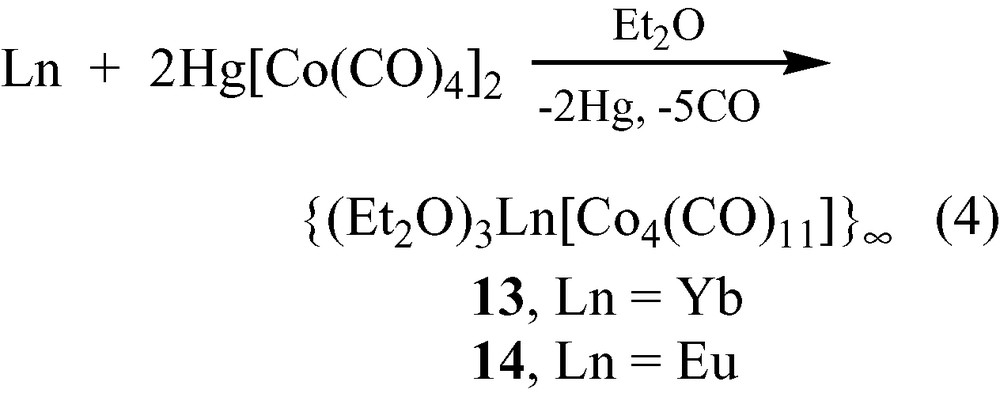
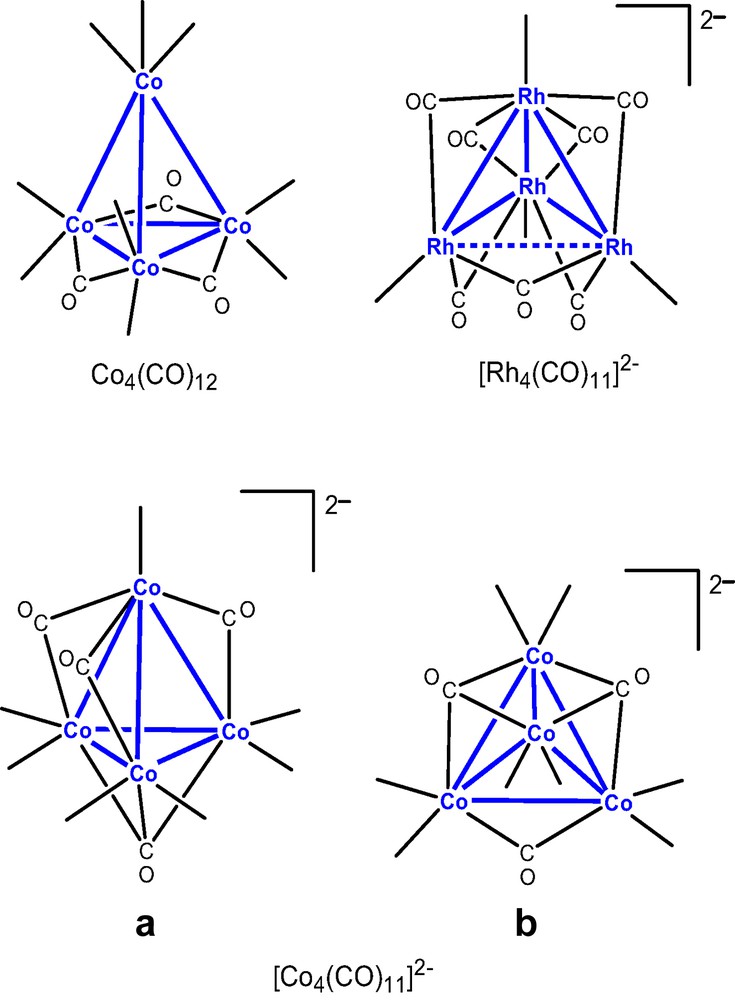
Even though the metal exchange process is similar to the reactions in strong coordinating solvents (Eq. (1)) in which Ln(0) is oxidized to Ln(II) and Hg(II) is reduced to Hg(0), the oxidation of Co in [Co(CO)4]– anions (Co [1–] to Co1/2–) and condensation of the monomeric units yield the tetracobalt undecacarbonyl dianionic cluster [Co4(CO)11] [2–], a cluster that had not been previously observed. Unlike reactions that produce ion pairs, 2-D heterometallic sheets supported by isocarbonyl bridges between Ln(II) and [Co4(CO)11]2– were generated in products 13 and 14. Compounds 13 and 14 are isomorphous.
The result of formation of isocarbonyl linkages is a 2-D polymeric sheet (Fig. 2a) that has two distinct building blocks. ‘Four-membered’ rings, with two Ln(II) centers and two [Co4(CO)11] [2–] clusters, are constructed with edge-bridging isocarbonyl bonds (Fig. 2b). The corner of the diamond-shaped ring is formed by the O–Ln–O angle that is almost 90°. Larger ‘eight-membered’ rings with four Yb(II) centers and four [Co4(CO)11] [2–] clusters have alternating face- and edge-bridging isocarbonyls bonds (Fig. 2c). From the top view of the two-dimensional network (Fig. 2a), the ‘eight-membered’ rings are oval shaped with two orientations. Cross-sections of the structure reveal a puckered sheet.
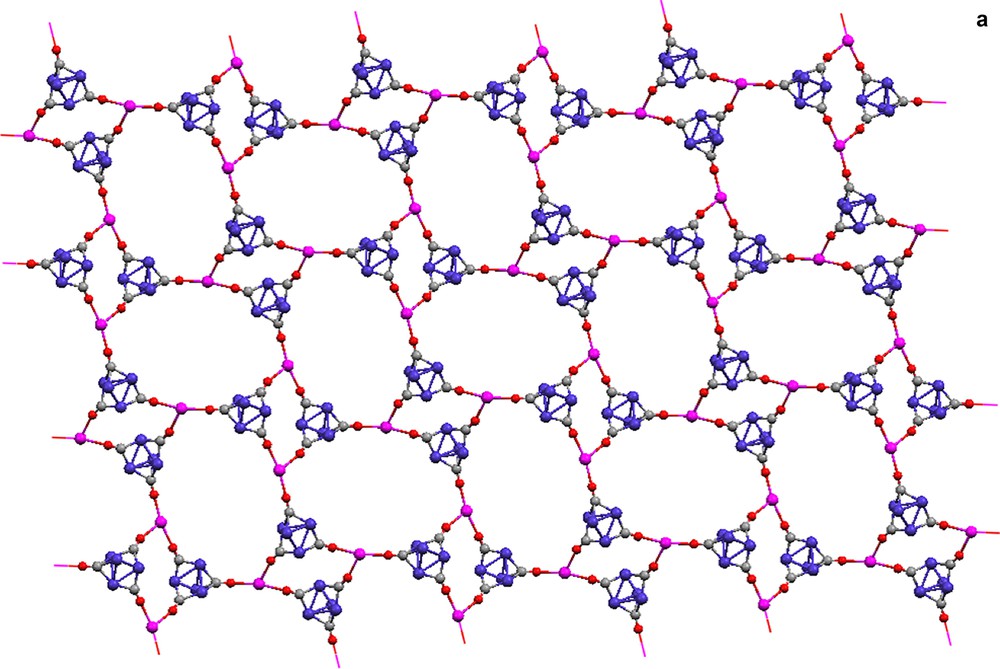


Molecular structures of the 2-D extended array of {(Et2O)3Yb[Co4(CO)11]}∞ (13). (a) Top view of the polymeric sheet, (b) the ‘4-membered’ ring building block, (c) the ‘8-membered’ ring building block, (d) and (e) cross-sectional views of the polymeric sheet. Et2O ligands are omitted for clarity. Terminal and μ2-carbonyls are represented as capped sticks.
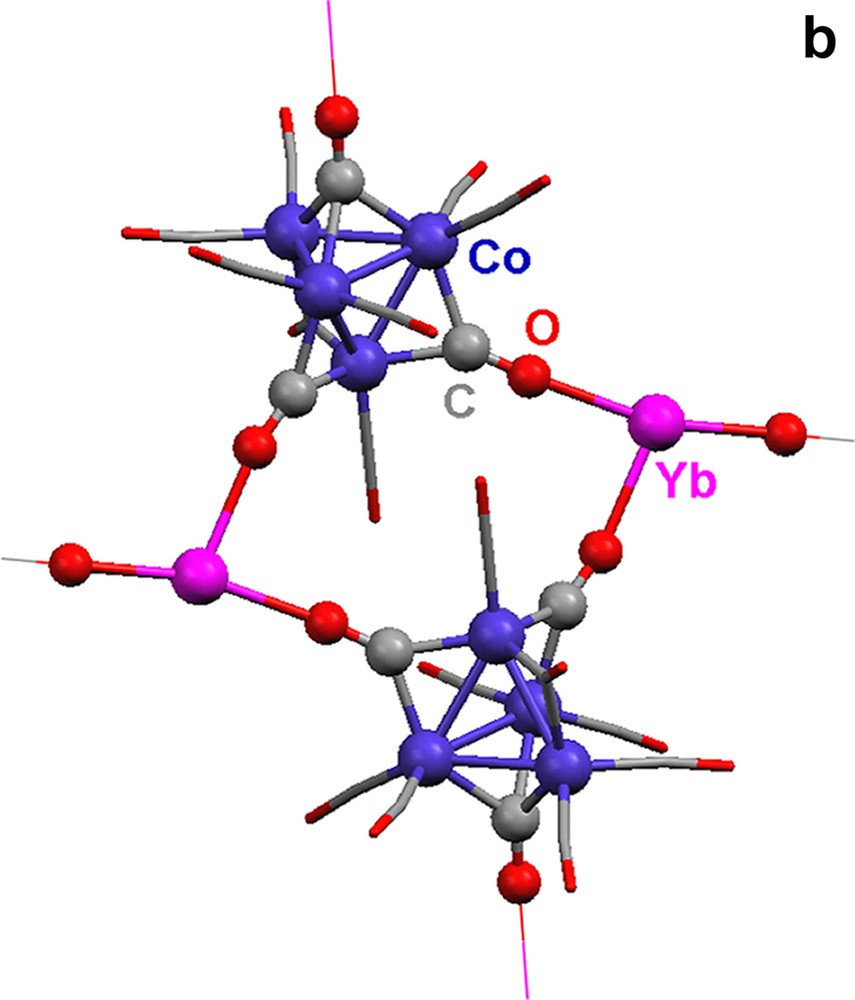
The tetrahedral tetracobalt dianionic cluster [Co4(CO)11]2– contains two distinct cobalt environments (Fig. 3a). The apical cobalt is bound to one terminal and three edge-bridging carbonyls, and the three basal cobalt atoms are each connected to one face-bridging, one edge-bridging, and two terminal carbonyls. Isocarbonyl linkages exist between Yb(II) and the face- and two of the three edge-bridging carbonyls. Terminal and one of the three edge-bridging carbonyls do not engage in isocarbonyl interactions. The three isocarbonyl linkages define the corners of a distorted tetrahedron with C3v symmetry (Fig. 3b, the bottom corner is the face bridging carbonyl which binds to a Yb(II) atom). The sheet is caused by the linkages from the cluster, i.e.: three corners of the tetrahedron. The Ln(II) atoms are 6-coordinate, with three solvent ligands and three isocarbonyl oxygens arranged as the corners of a distorted octahedron.
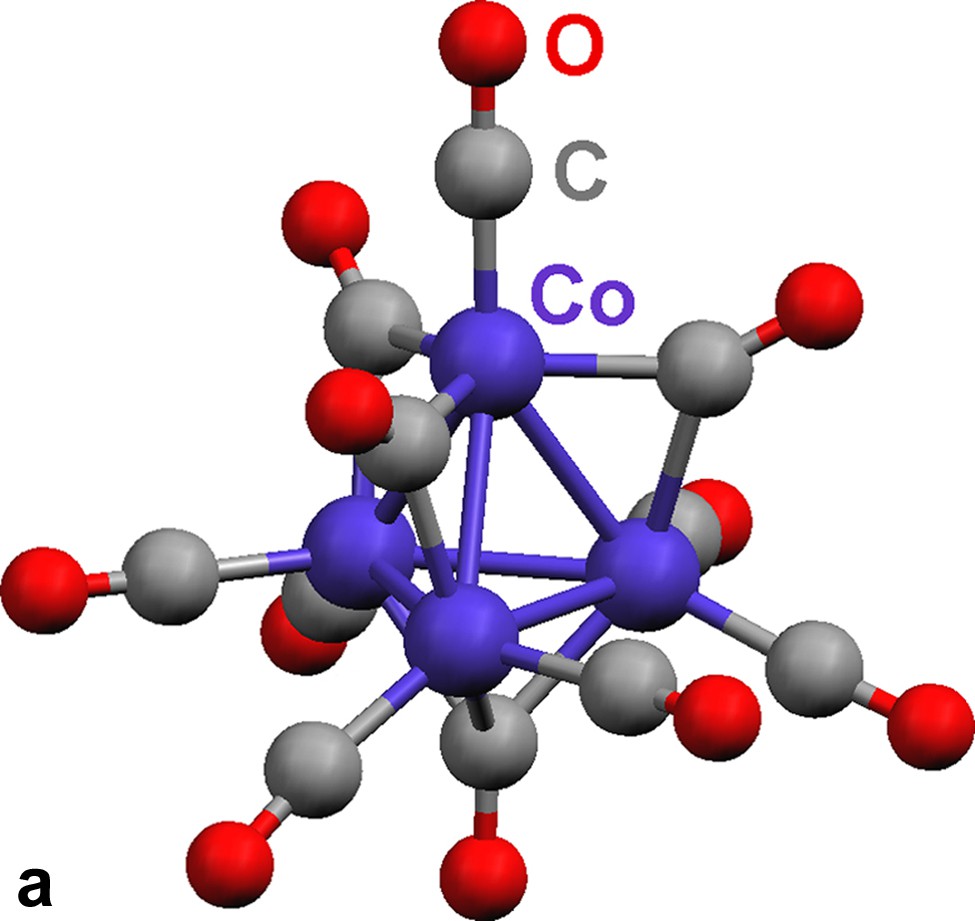
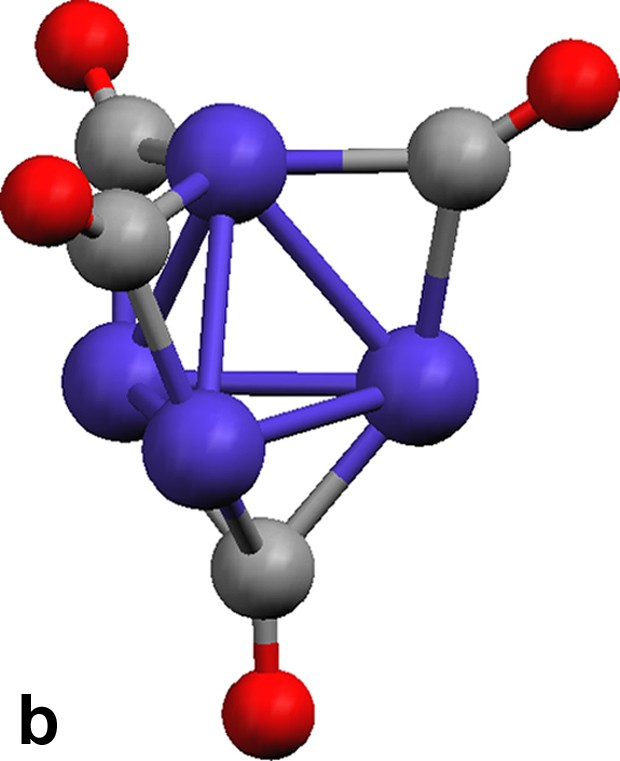
(a) Molecular structure of cluster [Co4(CO)11]2– in {(Et2O)3Yb[Co4(CO)11]}∞ (13). (b) Geometry of the [Co4(CO)11]2– cluster. Terminal carbonyls are omitted for clarity.
2.3.2 Conversion of ion pair complexes in a weakly nucleophilic solvent
We have demonstrated that solvent-separated products formed from weak carbonylate nucleophiles may be easily converted into isocarbonyls utilizing a weakly nucleophilic solvent [14].
2.3.2.1 Dissolution of [Ln(THF)x] [Co(CO)4]2 in Et2O
Formation of {(Et2O)2(THF)Yb[Co4(CO)11]}∞ (15). The ion-paired salt [Yb(THF)6] [Co(CO)4]2 (1) was stirred in Et2O for several days, the volatiles were removed, and the mixture was stirred in fresh Et2O for a day (Eq. (5)). The latter two steps were repeated. Filtration of the red–brown colored solution, and slow evaporation of the solvent produced crystals of [(Et2O)2(THF)Yb{(η2,μ4-CO)(η2,μ3-CO)2Co4(μ2-CO)(CO)7}]∞, 15 [17]. Compound 15 belongs to a different crystal system, but its structure is related to those of 13 and 14.
While 13 and 14 contain three THF molecules coordinated to Yb(II) and 15 contains one THF and two diethylether molecules coordinated to Yb(II), their two-dimensional structures are almost identical.
Formation of {(THF)5Eu[Co4(CO)11]}∞ (16). In a slightly different procedure from that described above, [Eu(THF)x] [Co(CO)4]2 was stirred in Et2O for several days and much of it remained undissolved ([Eu(THF)x] [Co(CO)4]2 which is less soluble in Et2O than [Yb(THF)6] [Co(CO)4]2). A minor quantity did react (Eq. (5)), and the resulting red colored solution was filtered and crystals of [(THF)5Eu{(η2,μ4-CO)2Co4(μ2-CO)(CO)8}]∞, 16, were isolated [17].
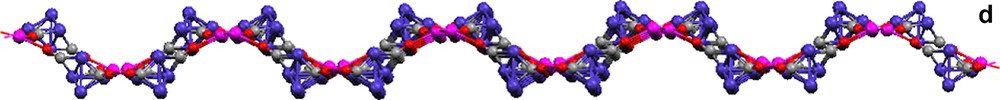
Complex 16 is composed of a [Co4(CO)11] [2–] cluster with C2v symmetry that coordinates to Eu(II) through isocarbonyl linkages to form a 1-D chain (Fig. 4).
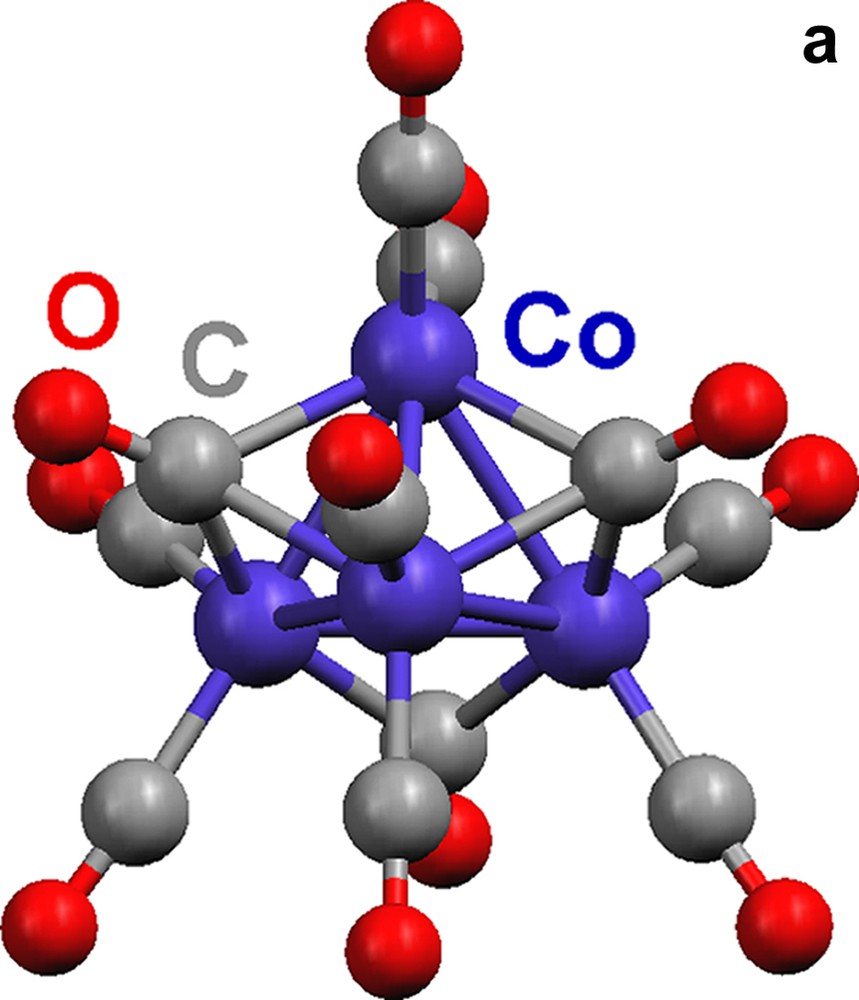
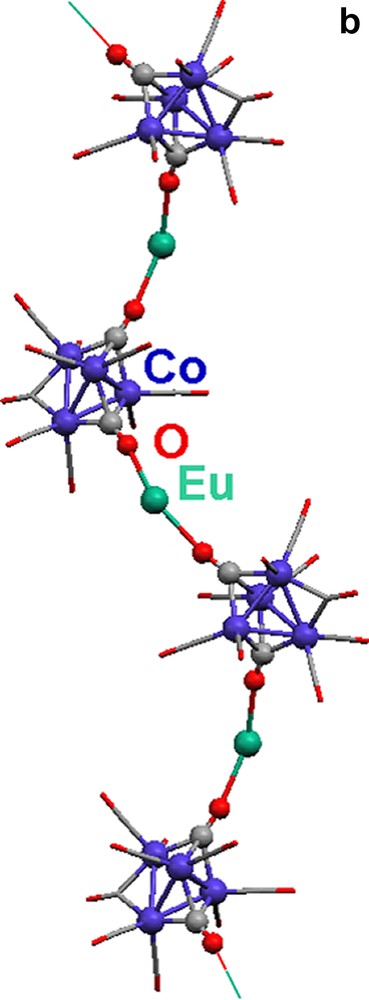
Molecular structures of {(THF)5Eu[Co4(CO)11]}∞ (16). (a) The [Co4(CO)11]2– cluster. (b) A portion of the 1-D zigzag chain (THF ligands are omitted; terminal and edge-bridging carbonyls are represented as capped sticks).
There are two face-capping, one edge-bridging, and eight terminal carbonyls in the cluster, [Co4(CO)11]2–. Isocarbonyl linkages (η2,μ4-CO) occur only through the two face-bridging carbonyls (Fig. 4a). The outcome of the oxymethylidyne linkages is a 1-D zigzag chain (Fig. 4b). The Eu(II) center is 7-coordinate, with five THF ligands and two isocarbonyl oxygens arranged in a distorted pentagonal bipyramidal geometry.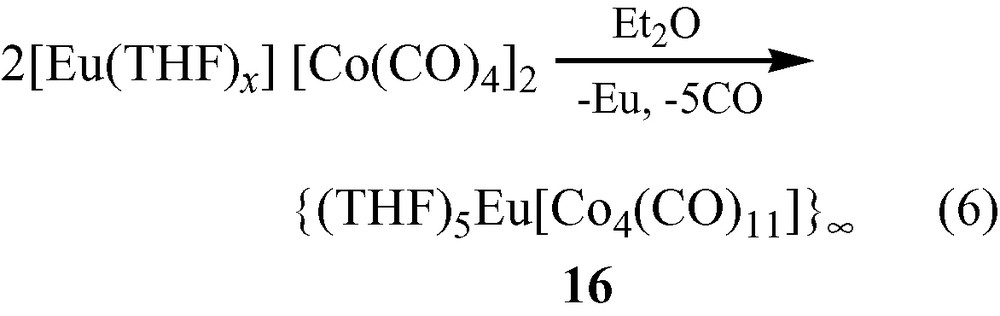
The [Co4(CO)11]2– cluster in 1–3 with one face- and three edge-bridging carbonyls (Fig. 3a) and the cluster in 4 possess one edge- and two face-bridging carbonyls (Fig. 4a) are isomers (A and B in Chart 3). They are isoelectronic with the neutral cluster Co4(CO)12, and [Rh4(CO)11]2– [18]. Note that [Rh4(CO)11]2– has no face capping carbonyls, only and more edge bridging carbonyls.
2.3.2.2 Dissolution of [Ln(THF)x] [Co(CO)4]2 in toluene
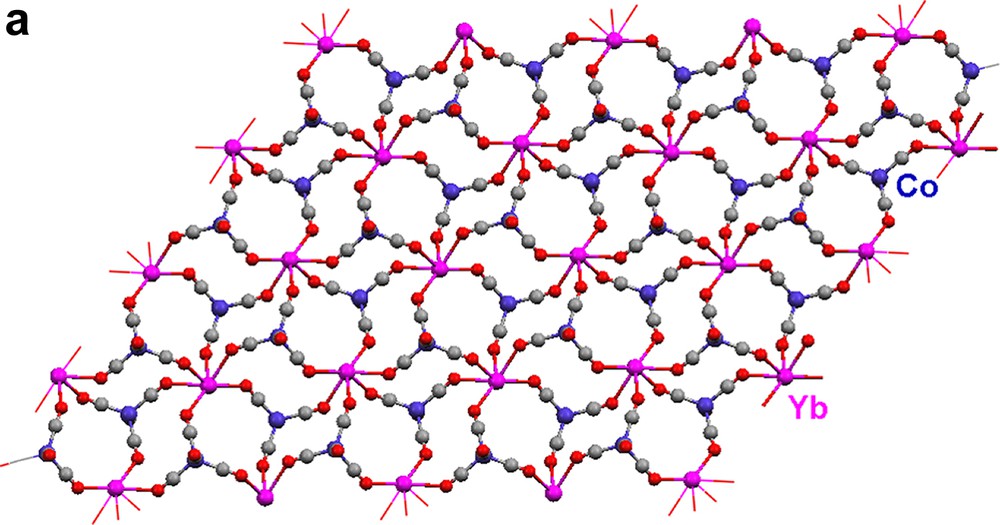
When solvent separated ion-paired salts 1 and 2 are dissolved in toluene, the neutral isocarbonyl polymeric array complexes, [(Pyr)4Yb{(μ-CO)2Co(CO)2}2]∞ (17) and [(THF)2Yb{(μ-CO)3Co(CO)}2·Tol]∞ (18) (Eqs. (7) and (8)) are produced [14]. Without the presence of other Lewis bases, the carbonyl oxygens can ligate to the Lewis acidic Yb2+ cations.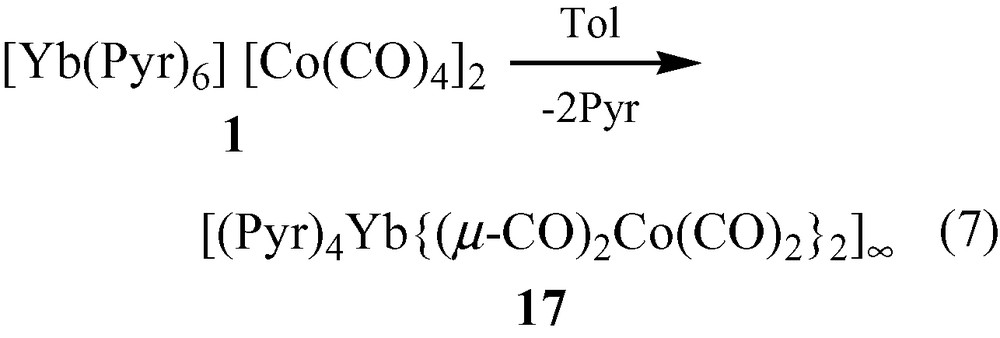
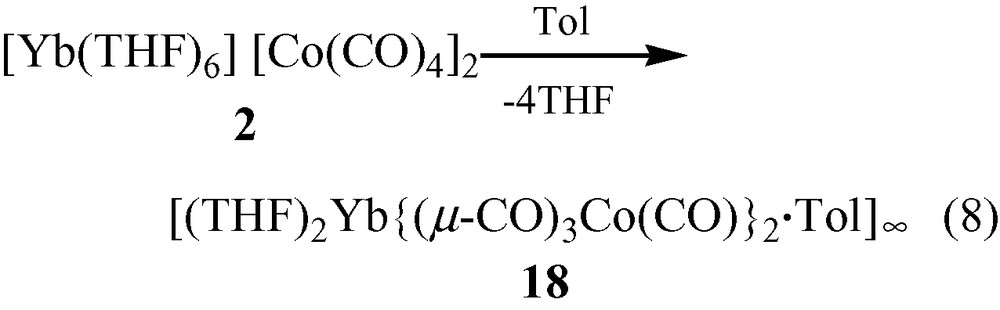
Each {Co(CO)4} moiety in 17 bridges through two carbonyls to two Yb(II) atoms (Fig. 5a). The Yb atoms, in turn, are bonded to four isocarbonyl oxygens. The ‘eight-membered’ ring building blocks (four Yb atoms on corners, four Co(CO)4 moieties on edges) generate a two-dimensional layer. The cross-sections of the structure (Fig. 5b, c) reveal a puckered sheet. Complexes 17 and 18 are formed under the same reaction conditions, but the structures of the products are in sharp contrast due to the fact that the starting materials differ in the ligands coordinated to the Yb(II) cation. Three carbonyls on each {Co(CO)4} unit in 18 are involved in an isocarbonyl interaction, and there are six isocarbonyl connections on each Yb(II) (Fig. 6a). The ‘four-membered’ ring basic unit (Yb atoms and two Co atoms) are incorporated into the sheet of 18. The consequence of these Yb–OC–Co linkages is a 2-D sheetlike puckered array (Fig. 6b). Complexes 17 and 18 belong to a rare class of Ln–M carbonyl extended polymeric structures, only three others of which have been structurally characterized (17, 18, and one prepared by Boncella and Andersen [7d]).
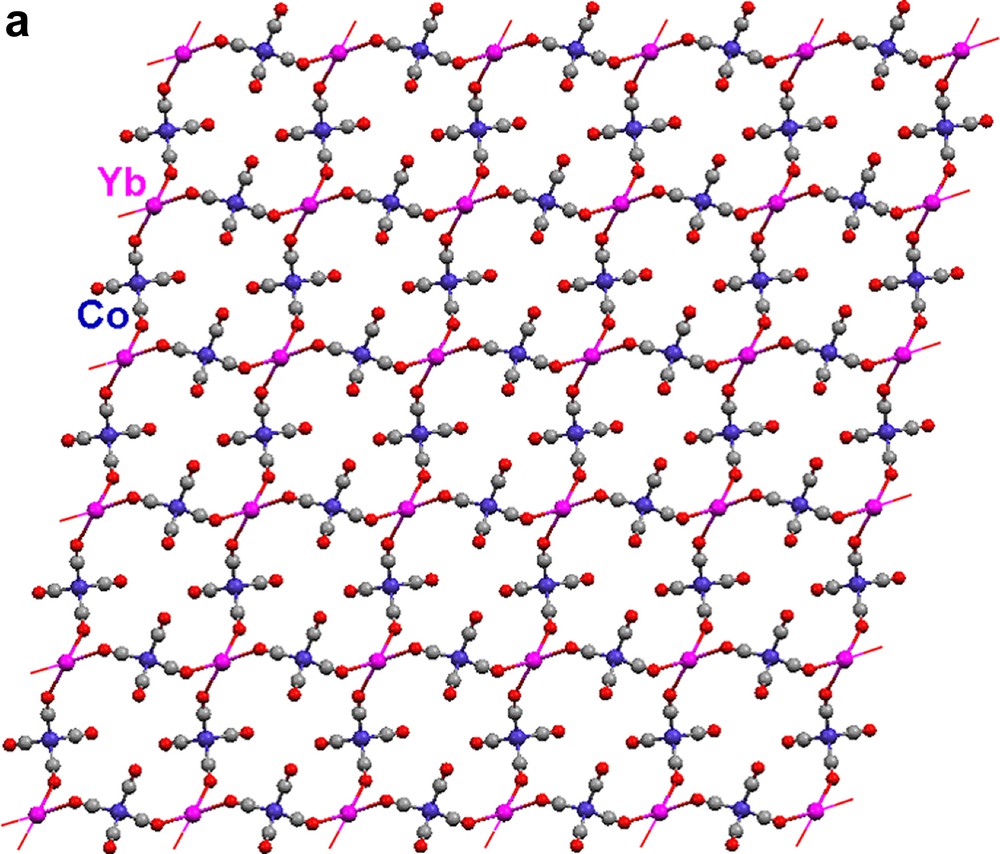
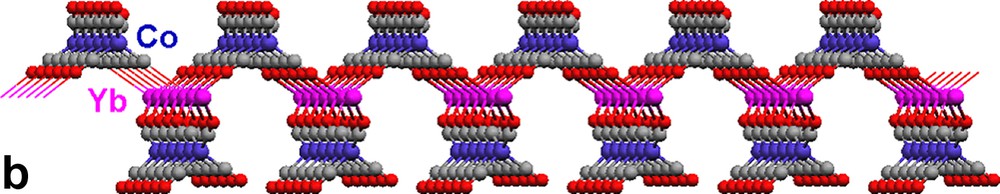
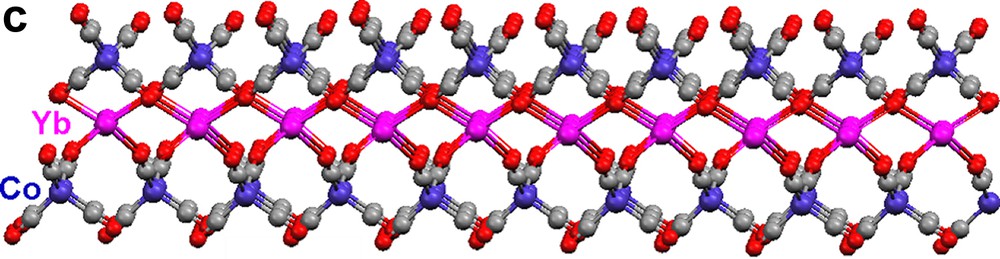
Molecular structure of 2-D polymeric array of [(Pyr)4Yb{(μ-CO)2Co(CO)2}2]∞ (17): (a) top view, (b) and (c) cross-sections. Pyridine ligands are omitted for clarity.
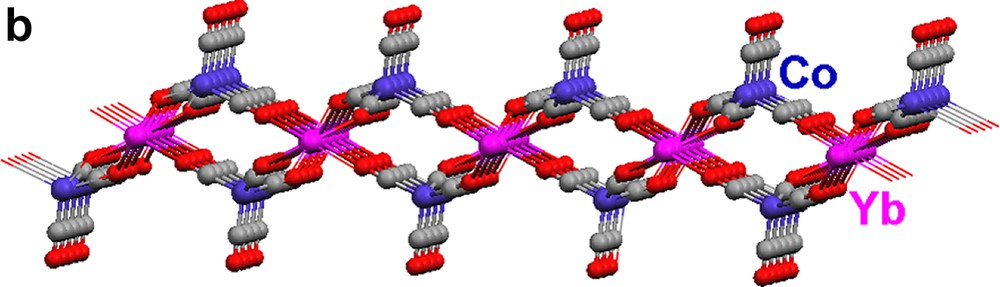
Molecular structure of the 2-D polymeric array of [(THF)2Yb{(μ-CO)3Co(CO)}2 ·Tol]∞ (18): (a) top view and (b) cross-section. THF ligands and the toluene of crystallization are omitted for clarity.
The carbonyl linkages in 17 and 18 are relatively weak. They are cleaved by pyridine and THF to regenerate the parent solvent-separated ions, 1 and 2. Compound 17 is not converted back to 1 completely because the solvent-separated ions exist in equilibrium with the weak contact ion pair and an isocarbonyl species.
2.4 Metal–metal bonded compounds
‘Supernucleophile’ [Fe(CO)4] [2] is the preferred reagent for designing Ln–M bonded products. By linking this anion with Yb(II), we and Beletskaya were the first to synthesize and structurally characterize (by single-crystal X-ray diffraction) Ln–M bonded systems [7h,10,11]. However, an actinide-transition metal complexes was prepared earlier by Sternal et al. [19].
Reduction of Fe3(CO)12 with three equivalents of Yb metal in liquid ammonia produces (NH3)2YbFe(CO)4 (Scheme 1) [10,11]. Dissolving (NH3)2YbFe(CO)4 in CH3CN causes the displacement of ammonia. Crystal structure analysis of the product, {[(CH3CN)3YbFe(CO)4]2·CH3CN}∞ (19), reveals a 1-D polymeric ladder that is constructed by the combination of Yb–Fe interactions and isocarbonyl linkages (Fig. 7). Two carbonyls on each {Fe(CO)4} unit participate in these isocarbonyl joints, which form ···Yb–OC–Fe–CO–Yb ··· zigzag chains. Ytterbium–iron bonds couple the chains together, and they constitute the rungs of the ladder. In a slightly modified procedure, {(CH3CN)3YbFe(CO)4}∞ (20) is isolated by quenching the (NH3)2YbFe(CO)4 reaction with CH3CN, removing the vocatives, and redissolving the solid in CH3CN (Scheme 1). Compound 20's structure is related to that of 19, but additional carbonyl bridges between the ladders create a 2-D sheet (Fig. 8; three carbonyls on each {Fe(CO)4} moiety engage in isocarbonyl interactions). From cross-sectional views (Fig. 8b, c), the 2-D sheet of 20 is puckered. Complex 20 has the structural feature (ladder) of 19 from the cross-sectional view of Fig. 8b. The most salient characteristics in the structures of 19 and 20 are the Yb–Fe bonds, which measure 3.010(1) and 3.046(1) Å, respectively. The bonds are comparable in length to those in YbFe2 alloy (3.00 Å) and are shorter than the sum of the metallic radii (3.2 Å). An interesting facet of the production of 1 and 2 is that Yb initially reduces Fe to form of [Fe(CO)4]2–, which then serves as a nucleophile to produce the Yb–Fe dative bond. It should be noted that without isocarbonyl linkages, complexes 19 and 20 would be a complex which is metal–metal bonded discrete molecule. It is the isocarbonyl interactions which build up 1-D ladder and 2-D sheets.

1-D polymeric ladder of {[(CH3CN)3YbFe(CO)4]2·CH3CN}∞ (19). CH3CN molecules are omitted for clarity.
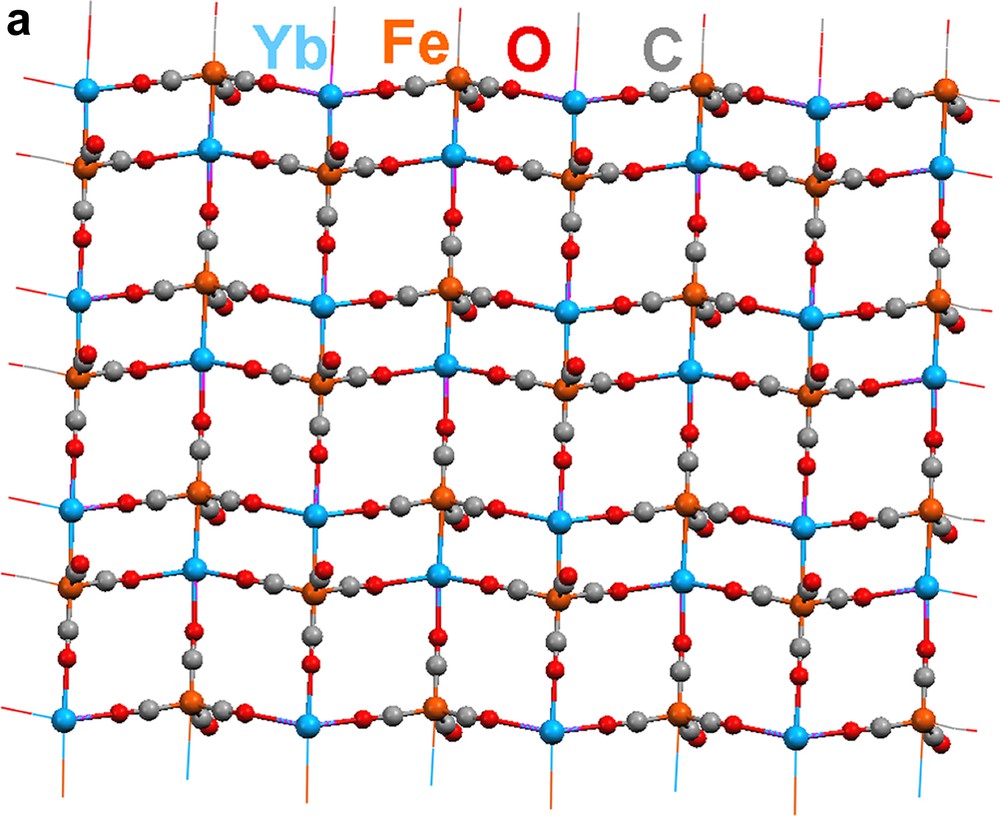
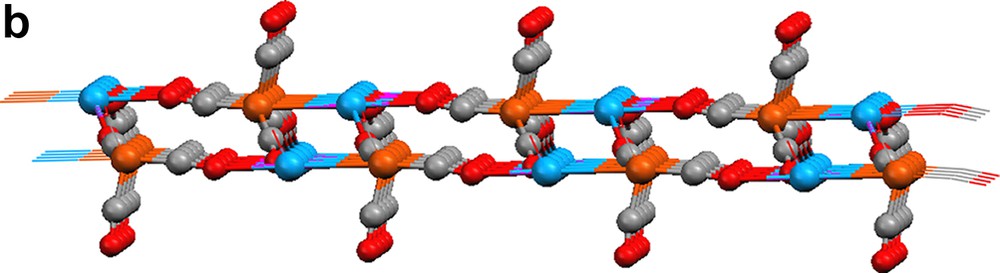
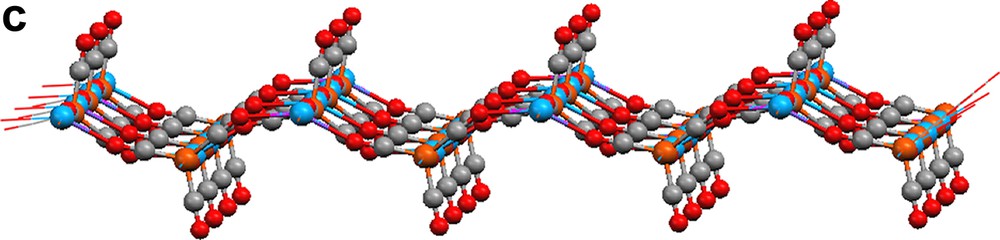
2-D extended sheet of {(CH3CN)3YbFe(CO)4}∞ (20): (a) top view, (b) and (c) cross-sections. CH3CN ligands are omitted for clarity.
3 Conclusions and outlook
In this article, we outlined our work on lanthanide–transition metal carbonyl systems. Our synthetic study demonstrates general synthetic procedures like transmetalation, M–M bond cleavage through lanthanide metal reduction in liquid ammonia or lanthanide/mercury amalgam reduction as facile and successful routes to prepare lanthanide–transition metal heterometallic complexes. Three kinds of interactions, ion paired, isocarbonyl linked, and metal–metal bonded complexes are observed. Relative nucleophilicities of [M(CO)y]n– and solvent play key rules in the formation of complexes with different Ln–M interactions. Structures span discrete molecules, 1-D, and 2-D arrays. Two isomers of the new [Co4(CO)11]2– cluster were obtained in the isocarbonyl complexes. All of the beautiful architectures are generated through isocarbonyl interactions between lanthanide and transition metal atoms. These linkages can be cleaved by relatively strong nucleophiles (including solvents) and that is that advantage for promotion in materials. As indicated in Section 1, heterogeneous catalysis is one of the potential applications we have been working and additional are under investigation.
Acknowledgements
This work was supported by the National Science Foundation through grants CHE99-01115 and CHE02-13491.