

1 Introduction
In order to attain a true microscopic understanding of kinetic mechanisms in heterogeneous (photo)catalysis, it is necessary to have a picture, as detailed as possible, of the nature of adsorption sites at the surface of the catalyst. Among the fields that would benefit from detailed adsorption studies at the semiconductor/solution interface, heterogeneous photocatalysis (applied both to synthesis and to water decontamination) [1,2] and dye photosensitization [2] should be mentioned. In fact, in the former, adsorption of the species to be oxidized (or reduced) affects the yield of the process and even the chemical path being favored by the photocatalyst. In the latter, studies on the adsorption geometry of the dye seem to be necessary to understand at a fundamental level the charge transfer process. The characterization of the adsorption geometry for different model compounds already studied from a kinetic viewpoint is also important. Among the different spectroscopic tools giving adsorbate vibrational information, laser Raman scattering techniques [3] present some advantages, deriving in part from the low Raman cross section of water: they can provide spectra for adsorbates in a single scan, possess a high spectral resolution and can probe an interface between two dense media. It is also remarkable that Raman spectroscopy is a relatively simple optical technique that can be easily coupled to different experimental set-ups and samples. However, the use of Raman spectroscopy for studying adsorption is limited by its low sensitivity, related to the second order nature of the Raman effect. Two approaches have been employed in the past to overcome this difficulty: resonance Raman spectroscopy (RRS) and surface enhanced Raman spectroscopy (SERS) [3].
Resonance Raman occurs when the scattering molecule absorbs incident radiation (i.e. when the laser beam excites electronically the molecule), which leads to intense Raman spectra. This enhancement is not restricted to adsorbed molecules, rendering difficult the study of reversible adsorption because of interference from molecules in the bulk. If the adsorption is almost irreversible (high adsorption equilibrium constant), interference from dissolved species can be neglected. Of course, RRS can be in addition specific to the interface if absorption of exciting radiation occurs only in the adsorbed state. A few RRS studies on the adsorption of different species at the surface of titanium oxide have appeared in the past. Already in 1981 Brazdil and Yeager [4] studied the adsorption of two molecules giving resonance (p-nitrosodimethylaniline and p-dimethylaminoazo-benzene) on some semiconducting oxides in contact with a gas phase. In the solid/liquid interface, the adsorption of sensitizing dyes and ferrocyanide has been also dealt with [5–12].
On the other hand, SERS constitutes another approach for enhancing the typically weak Raman intensities [13]. The enhancement appears in the interphasial region of roughened samples of certain metals (Cu, Ag and Au). Two mechanisms seem to contribute to the enhancement: the so-called chemical and the electromagnetic. While the first is limited to molecules directly adsorbed at the metal surface, the second extends spatially out of the metal surface. However this extension is quite limited, i.e. the enhancement decreases rapidly with distance. The question of the existence of SERS effect on pure, roughened semiconductor samples is still controversial. Quagliano et al. [14,15] have claimed the existence of SERS for the etched GaAs/Rubpy system but the use of a dye molecule as a probe adds some uncertainty to this conclusion as the enhanced signals could be due to the combination of a large internal surface area and a resonant effect in the adsorbed molecule. Nevertheless, Raman spectra for pyridine adsorbed on III–V semiconductor quantum dots have been reported very recently [16]. Certainly, enhancement can be achieved by adding Ag nanoparticles (or depositing Ag islands) to the semiconductor sample [14,15,17–19] but this approach suffers from several drawbacks. On the one hand, the adsorptive and catalytic capabilities of the sample result seriously affected: instead of a semiconductor nanostructured sample, a composite is being studied. On the other, there are no clear criteria to decide whether the detected signals are coming from the semiconductor or contrarily coming from the silver surface. It should be borne in mind that the enhancement is expected to be more important for molecules directly adsorbed on silver. The possibility exists of isolating the Ag particles from the solution with covering them by a layer of the adsorbent oxide being studied. Although examples of such a tactic are sparse, it is remarkable that adsorption of species from solution has been examined in the case of SiO2-overcoated silver-island films [20,21].
This paper is organized as follows. We begin with the possibility of applying SERS to nanoporous thin films (electrodes) of anatase deposited on roughened gold samples by taking advantage of the spatial extension of the electromagnetic enhancement. We pay attention to the possible interference of the gold substrate and explain how to deal with it. For validating our approach we review briefly the adsorption of phthalic acid on anatase [22] and then extend this study to methanol and formic acid. For the latter molecules we correlate the spectroscopic results with previous photoelectrochemical studies on their photooxidation [23]. The second part of this article is devoted to present some preliminary results of the application of resonance scattering to an adsorbate that is typically the subject of photocatalytic fundamental studies such as salicylic [24–26] and to an adsorbate used as a sensitizing modifier of TiO2, such as ascorbic acid [27,28]. These adsorption studies can be applied both to concentrated slurries of anatase and to thin films deposited on conducting glass.
2 Experimental
For the SERS-based experiments, nanoporous TiO2 films were coated on previously roughened gold samples (by applying 25 oxidation–reduction cycles as in [29]) through deposition of 3 μl of a 15.6 mM anatase suspension (Alfa Aesar, APS 30 nm) that was allowed to dry in air at room temperature. Films were also prepared by electrophoresis in a TiO2 suspension (1% w/v in ultrapure water). A bias of 10 V was applied between the gold electrode (previously roughened) and a gold counterelectrode for 5 s. The film so formed was allowed to dry in air, as in the previous procedure. Once the anatase thin film was deposited onto the roughened gold surface, the sheathed gold rod was mounted on an electrochemical PTFE cell designed to acquire in situ Raman spectra [30]. A fused silica window separated the microscope objective from the electrolytic solution, which was 0.1 M HClO4 (Merck p.a.) and 2 mM phthalic acid (potassium hydrogen phthalate Merck R.P. Normapur A.R.). A Pt wire was used as counter electrode, whereas a saturated calomel electrode (SCE) was used as reference electrode contacting the working solution through a Luggin capillary. All the potential values are referred to the SCE.
For the RRS experiments, thin films of anatase (a few μm in thickness) were prepared on a conducting glass substrate (F-doped SnO2, FTO, Asahi Glass Co, Japan) by electrophoresis in a TiO2 suspension (1% w/v in methanol). A bias of 10 V was applied between the FTO electrode and a gold counterelectrode for 1 min. The films so formed were allowed to dry at room temperature, before thermal treatment at 450 °C in air during 1 h. Raman experiments were also performed with concentrated anatase slurries (0.17 g cm–3) in 15 mM salicylic (for synthesis, MERCK) or l-(+)-ascorbic acids (Panreac) + 0.1 M HClO4 solution. A thin layer configuration cell made of stainless steel and PTFE, equipped with a fused silica window was used for the experiments with the slurries.
Raman spectra were obtained with a LabRam spectrometer (from Jobin-Yvon Horiba). The typical slit and pinhole employed were 200 and 600 μm, respectively. The excitation line was provided either by a 17 mW He–Ne laser at 632.8 nm or by a 50 mW Ar-ion laser at 514.5 nm. The laser beam was focused through a 50× long-working distance objective (0.5 NA) into a 2-μm spot at the electrode surface. The spectrometer resolution was better than 2–3 cm–1 and the detector was a Peltier-cooled charge-couple device (CCD) (1064 × 256 pixels). Unless otherwise indicated, all the spectra are shown as obtained.
3 Results and discussion
3.1 SERS applied to adsorption studies on nanoporous anatase thin films (electrodes)
Except for a very recent report by Quagliano [16] dealing with III–V quantum dots, semiconductor nanocrystals do not seem to sustain a significant SERS effect. As mentioned above the obtainment of spectra for adsorbates requires the introduction of one of the coinage metals (most frequently silver) in the nanostructured semiconductor sample, but this approach alters, sometimes dramatically, the properties of the catalyst (adsorbent). We have developed an approach that permits the acquisition of spectra for adsorbates avoiding interference from the gold substrate. The application of an external bias allows the use of the nanoporous semiconductor film as an electrode [22]. Its validity was tested by studying the adsorption of phthalate.
Our procedure relies on the deposition of a thin anatase layer onto a roughened gold sample, active as a SERS substrate. We have studied first the adsorption of phthalic acid on the bare Au electrode and identify the potential region where the species is adsorbed by means of SERS. We found that adsorption occurs from around 0.4 to 1.0 V and that the main adsorbed species is phthalate, in spite of the fact that the solution is acidic (pH 1), being the protonated species predominant in solution. Desorption at less positive potentials is just a reflection that the monitored species is an anion whose adsorption is disfavored for less positive potentials. Desorption for potentials higher than 1.0 V is caused by the adsorption of oxygenated species (O/OH) on the gold surface, which leads to a displacement of other adsorbates. To avoid interference from the substrate we applied a potential outside the range mentioned above. Concretely a potential of 1.4 V was chosen. The corresponding SER spectrum is given in Fig. 1a. As observed, it is featureless, except for the broad, intense band at around 600 cm–1 due to the stretch of the metal–oxygen bond originating from the potential-induced adsorption of oxygenated species.
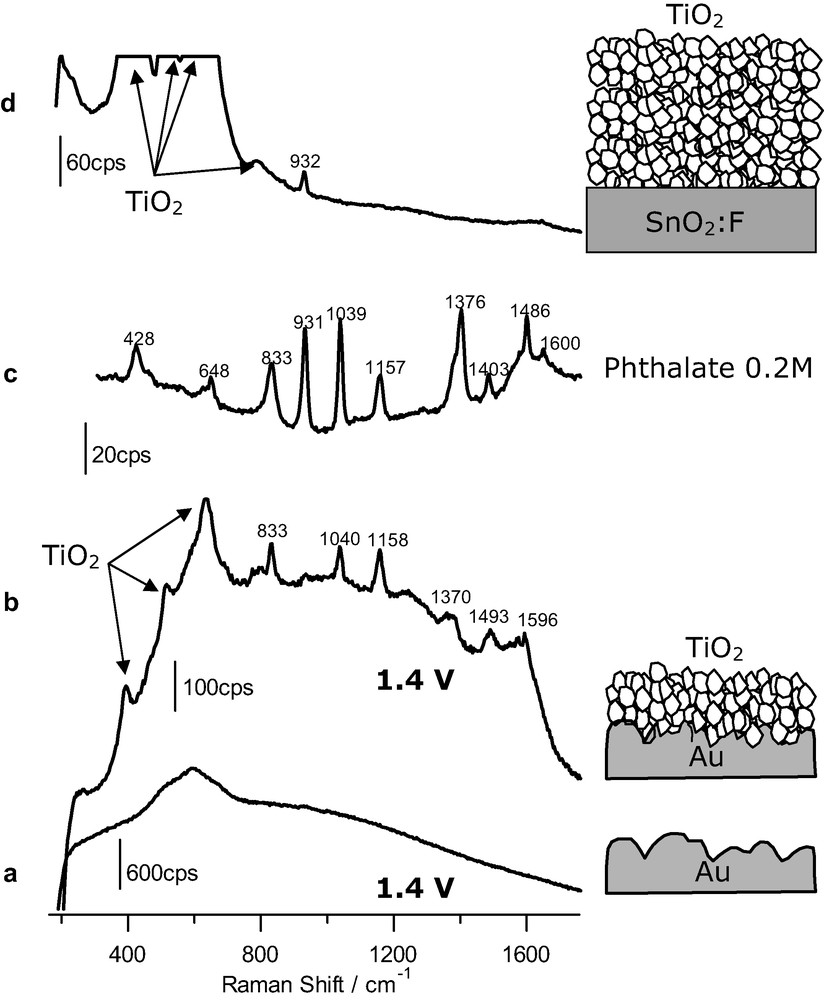
(a) SER spectrum obtained at 1.4 V for a gold electrode (acquisition time: 20 s) in the presence of 2 mM phthalic acid. (b) Same as for a but for a gold electrode covered with an anatase nanoparticulate thin film (time: 30 s). (c) Raman spectrum obtained for a 0.2 M phthalate solution, pH 12 (time: 120 s). (d) Raman spectrum obtained for a nanoporous film of anatase on FTO conducting glass (acquisition time: 120 s) in the presence of 1.5 mM phthalic acid. The excitation line was provided in all cases by a 17-mW He–Ne laser. Supporting electrolyte: HClO4 0.1 M.
As we wanted to study the adsorption on the semiconductor nanoporous film, we repeated the experiments, but covering this time the gold substrate with a thin layer of anatase nanoparticles. The resulting spectrum under polarization at 1.4 V is shown in Fig. 1b. One can realize bands due to vibrations of the bulk anatase, but also bands associated to vibrations of an adsorbed species (notably at 833, 1040, 1158, 1370, 1493 and 1596 cm–1). Fig. 1c shows the spectrum obtained for a 0.2 M solution of phthalate in an alkaline solution of sodium perchlorate (by addition of sodium hydroxide). Spectra in Fig. 1b, c are similar, which indicates that, as in the case of the bare gold substrate, the adsorbed species is phthalate. In this respect it is very informative the absence of the ν(C=O) band at around 1700 cm–1, typical of undissociated carboxylic acids. More details on these results and subsequent discussion can be found in [22].
A control experiment was performed to rule out the possibility that an anatase thin film could provide by its own any type of enhancement leading to spectra for adsorbates. Although a nanoporous thin film (around 2-μm thick and made of 30-nm-diameter nanoparticles) has a quite large inner surface area, in principle this is not enough to allow the obtainment of non-enhanced spectra for adsorbates. In this case a thin nanoporous anatase layer is deposited on FTO, which is not an active SERs substrate. The film is made thicker (by one order of magnitude) than when the roughened gold substrate was used, in order to increase the inner surface of the film and facilitate the eventual obtainment of adsorbate signals. Fig. 1d corresponds to the spectrum obtained in this case. As observed, no bands due to the presence of adsorbed phthalate can be distinguished, the only features in the spectrum corresponding to bulk water and perchlorate.
The experiments summarized in Fig. 1 evidence that surface enhanced Raman spectroscopy can be extended to adsorption studies on nanostructured semiconductor films. The basic idea underlying our approach is the existence of an electromagnetic enhancement due to a laser-induced plasmonic excitation of the rough gold sample [13]. The enhancement is not limited to the surface of the gold sample but extends spatially into the phase in its vicinity. In fact, this extension seems to be favored in the presence of the solution and semiconductor phases over the vacuum situation [31]. If there is an anatase nanoparticulate overlayer on the gold template, the bottommost part of this film contributes to the SER spectrum as observed in Fig. 1b. Not only the bands due to bulk anatase are enhanced but also those due to the species adsorbed on the anatase nanoparticles. As the anatase thin film is nanoporous, the solution phase is also in contact with the gold template, which in principle could be a source of interference. If adsorption of the species to be studied occurs on gold, an electrode potential should be applied to the gold substrate in order to avoid adsorption. This is achieved for most anionic and neutral species by applying potential values sufficiently low to induce the desorption of the anion or high enough to trigger the displacement of the adsorbed species by the adsorption on gold of OH and/or O species. The control experiment shown in Fig. 1d discards the possibility that the observation of bands due to the adsorbate could be caused solely by the existence of a high inner surface in the nanostructured film allowing for an increment in the number of sampled adsorbed molecules. In addition, the possible existence in this case of an enhancement due to resonant effects linked to the formation of a charge transfer complex between the Ti(IV) site and the adsorbate is discarded.
Recently, we have embarked on a project with the aim of discriminating between direct (via valence band holes) and indirect (via surface-bound hydroxyl radicals) hole transfer in heterogeneously photocatalyzed oxidation reactions of RH2 (RH2 representing any reduced dissolved species). In this context, we have developed a kinetic model that allows to specify the main mechanism of hole transfer on the basis of experimental measurements. A model for polycrystalline electrodes giving current doubling [32] has been proposed and discriminating tests have been developed [23]. The most useful one consists of studying the slope at the origin of the stationary photocurrent (plateau) versus RH2 concentration plot , as a function of the incident photon flux, . If the slope depends linearly on , direct photooxidation predominates, whereas, if the slope depends linearly on , the indirect pathway prevails. Photoelectrochemical measurements with a polycrystalline anatase electrode have evidenced that in acidic media the photooxidation of methanol seems to occur indirectly while that of formic acid appears to be direct.
As discussed previously [23] on the basis of electron transfer theories, in most cases, the direct attack of valence band holes will occur as long as the hole acceptor (species to be oxidized) is adsorbed at the semiconductor surface. In this way the hole can be captured inelastically in a non-activated, fast process by the adsorbed species, which can be considered as an extrinsic surface state. In the case of non-adsorbed RH2 species, the electron transfer should be isoenergetic, the direct hole capture occurring if the redox potential corresponding to the first electron exchange approaches that of the semiconductor conduction band edge. This is unlikely in the case of anatase as the latter value is very high. Better overlapping should characterize electron exchange with adsorbed hydroxyl species, whose redox potential is less positive than that of the conduction band edge.
As there seems to exist a close correlation between the adsorption of the species to be oxidized and the mechanism of hole transfer, it is interesting to apply SERS as explained above to the cases of methanol and formic acid and check if the results are compatible with our photoelectrochemical experiments [23].
As a reference, Fig. 2 shows the Raman spectra obtained for pure methanol and formic acid as well as for 1 M aqueous solutions in both alkaline and acidic solution. The changes observed in the spectra for the solution of formic acid depending on pH are a consequence of the predominance of undissociated formic acid at low pH values and of formate in alkaline solution.
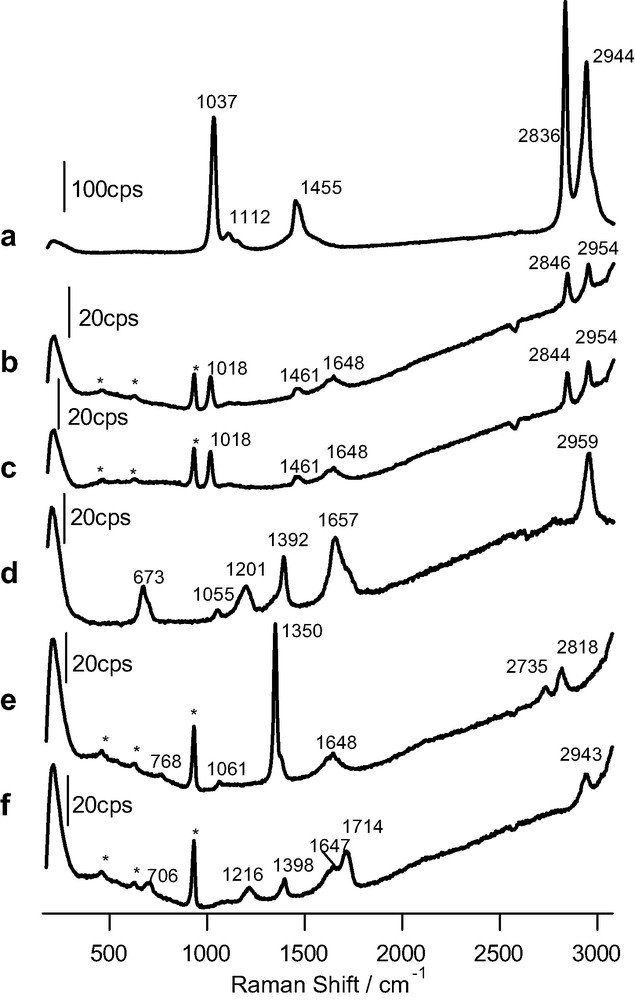
Raman spectra obtained for: (a) pure methanol (acquisition time: 20 s); (b) 1 M methanol solution (pH 12) in 0.1 M perchlorate (time: 120 s); (c) 1 M methanol solution (pH 1) in 0.1 M HClO4 (time: 120 s); (d) pure formic acid (time: 60 s); (e, f) as in b and c, but for formic acid. The excitation line was provided by a 17-mW He–Ne laser. Bands marked with an asterisk are due to bulk perchlorate.
Fig. 3 corresponds to SERS experiments performed with bare roughened gold and with an anatase overlayer deposited on roughened gold in contact with either formic acid (spectra a and b) or methanol (spectra c and d) dilute solutions in 0.1 M HClO4. The experiments are done in a spectroelectrochemical cell and a potential of 0.6 V (double-layer region of the gold electrode) was applied in all cases. As observed, spectra b and d in Fig. 3, obtained for bare gold, are virtually featureless, meaning that in these particular cases, no significant adsorption of the organics occurs even at double layer potentials such as 0.6 V. Therefore, we do not need to apply a potential leading to the oxidation of the gold substrate to avoid interference. Spectrum c obtained for an anatase overlayer is also rather featureless, pointing to the absence of significant specific adsorption of methanol on the anatase nanoparticles.
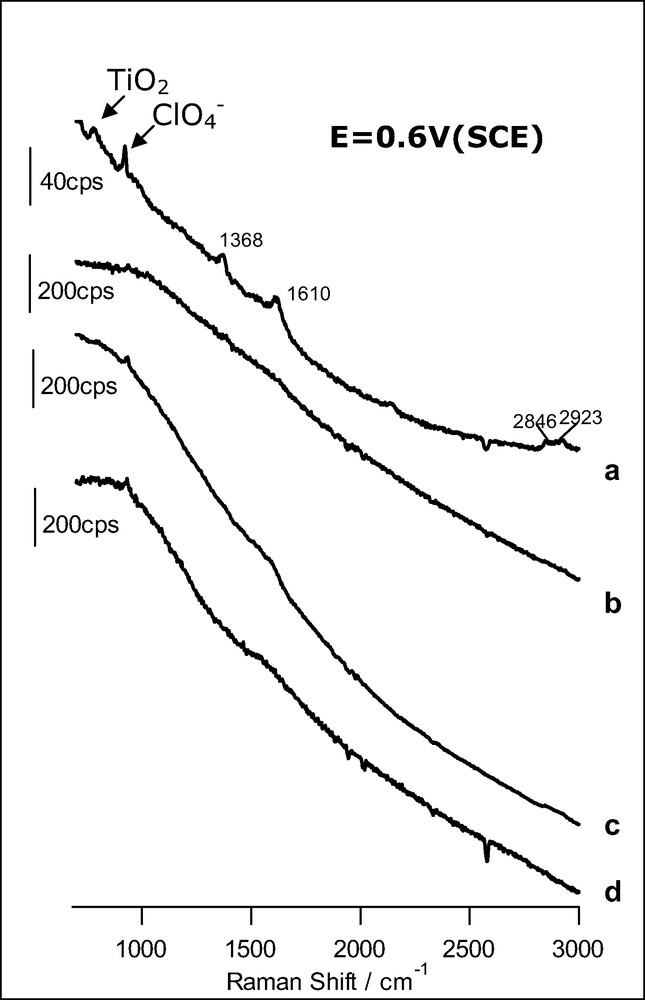
SER spectra obtained at 0.6 V (SCE) for a gold electrode: (a) covered with an anatase nanoparticulate thin film in the presence of 1 mM HCOOH (acquisition time: 60 s); (b) bare in the presence of 1 mM HCOOH (acquisition time: 10 s); (c) covered with an anatase nanoparticle layer in the presence of 1.5 mM CH3OH (time: 10 s); (d) bare in the presence of 1.5 mM CH3OH (time: 10 s). The excitation line was provided by a 17-mW He–Ne laser. Supporting electrolyte: HClO4 0.1 M.
Fig. 3a shows the spectrum corresponding to the anatase overlayer in contact with a formic acid solution. In clear contrast with spectrum b, bands at 1368, 1610, 2846 and 2923 cm–1 appear now, evidencing the adsorption of formic acid. The first two bands can be ascribed to the symmetric and asymmetric stretching of carboxylate, respectively. The two bands at higher wavenumbers correspond to C–H vibrations. Interestingly, the absence of the C=O stretching band (at around 1714 cm–1, see Fig. 2) discards the molecular, associative adsorption of formic acid. At the surface, the predominant species is formate, even at a pH as low as 1. A similar behavior has been revealed very recently by means of infrared spectroscopy for thin films of rutile nanoparticles [33].
The application of Raman spectroscopy to the adsorption of methanol and formic acid on anatase illustrates how the vibrational, microscopic information furnished by the technique serves to confirm mechanistic ideas on the photooxidation of these compounds on anatase linked to macroscopic photoelectrochemical measurements [23]. In fact, direct hole transfer photooxidation of formic acid deduced from kinetic electrochemical measurements was assumed to be linked to adsorption based on the theories of electron transfer and this adsorption could be confirmed by means of Raman spectroscopy.
3.2 RRS applied to adsorption studies on nanoporous thin films and slurries of anatase
Although, as mentioned above, resonant Raman studies at the SC/solution interface are far from new, until now the published reports have been restricted mainly to the phenomenon of dye sensitization. We present in the following preliminary results on the adsorption of relatively simple molecules, studied frequently in heterogeneous photocatalysis or, more broadly speaking in the field of photoelectrochemistry (charge transfer). Concretely we deal with the adsorption of ascorbic acid and salicylic acid on anatase nanoparticles. We take advantage of the formation, upon their adsorption at the acid sites of the oxide surface (Ti(IV) sites), of a charge transfer complex [27,28,34] absorbing in the visible (absorbing the laser exciting radiation) and thus allowing for a resonant effect. Of course, as these compounds do not absorb visible light when dissolved, no interference is produced by the solution phase: only the adsorbed molecules are sampled.
Fig. 4 shows Raman spectra corresponding to anatase concentrated suspensions in a 14.5 mM solution of either ascorbic or salicylic acids and 0.1 M HClO4. Inset shows a sketch of the cell used for these measurements. The suspension is enclosed between a fused silica window, a PTFE spacer (80 μm) and a bottom plate of the same material.
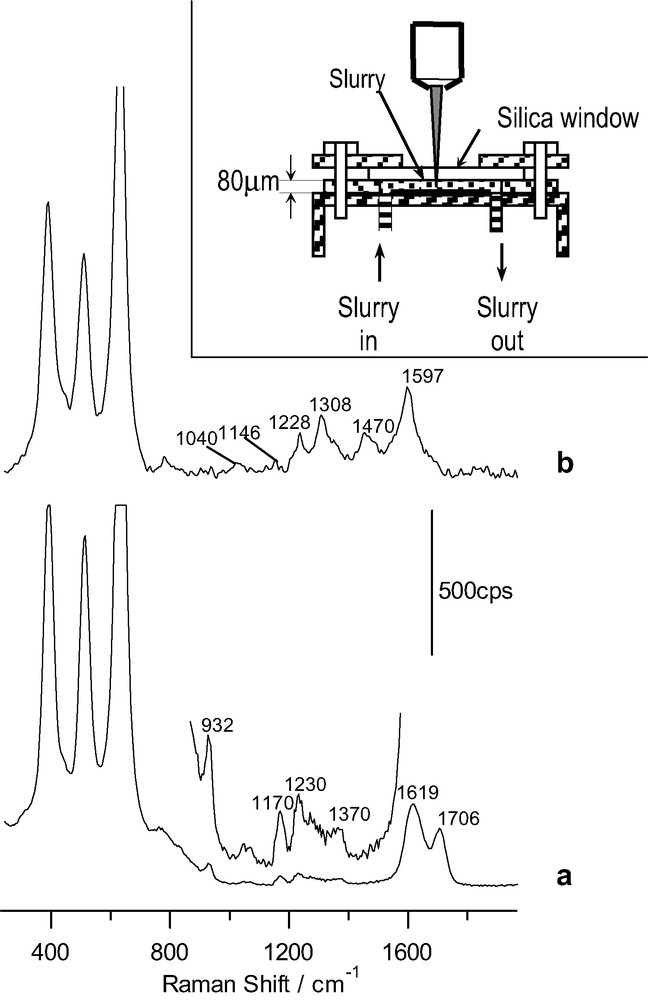
Raman spectra for a TiO2 slurry in presence of: (a) 14.5 mM ascorbic acid in 0.1 M HClO4 (acquisition time: 40 s); (b) 14.5 mM salicylic acid in 0.1 M HClO4 (time: 4 s). Inlet: sketch of the spectrochemical cell for the slurry analysis with the confocal microprobe Raman system. The excitation line was provided by an Ar+ laser.
Fig. 4a corresponds to the spectrum obtained for the anatase suspension in the ascorbic acid solution. It shows several features attributable to adsorbed ascorbic acid. Two intense bands appear at 1619 and 1706 cm–1. In addition weak bands appear at (1070), 1170, 1230 and 1370 cm–1 (the band at 932 cm–1 is due to bulk perchlorate). Although different assignments have been proposed for them [27,35–37], there is a higher degree of agreement for the most intense ones: that at 1619 cm–1 would correspond to ν(C=O) and that at 1706 cm–1 to (ν(C=O)). It is interesting to compare these frequencies with those appearing in ascorbic acid (1753 and 1672 cm–1, respectively). Both of them are shifted toward lower frequencies in the adsorbed species attaining values similar to those of sodium ascorbate [37]. This may be interpreted as a diminution in the electron density in the C=C and C=O bonds typical of charge delocalization among C1, C2 and C3 in ascorbate, which seems to be the predominant adsorbed species in spite of the low solution pH. Although on the basis of the acquired spectrum for the adsorbed species it is not possible to deduce the structure and mode of bonding of the adsorbed species, our results are compatible with previous results, suggesting a bidentate binding forming a 5-member ring around the surface Ti(IV) site [27,28]:
Fig. 4b corresponds to the spectrum obtained for an anatase suspension in a dilute salicylic acid solution. Different bands due to an adsorbate appear in the region 1000–1700 cm–1. Most of them have been assigned previously to C–C ring stretching vibrations (those at 1040, 1146, 1308, 1470 and 1597 cm–1) [38–40]. The band at 1228 cm–1 may be associated to the phenolic C–O stretching. The fact that it is shifted with respect to aqueous salicylate has been interpreted as a consequence of coordination through the phenolic group to the Ti(IV) surface site [38]. Again a chelate structure seems to be favored in agreement with previous reports on the adsorption of salicylate on different oxides [38,39]:
Let us examine finally the time evolution of the Raman spectra since in this case the electronic transition caused by absorption of the excitating radiation corresponds to a charge transfer from the excited molecule into TiO2 (provoking its sensitization) [27,34]. In other words, the electronic transition leads to a charge separation at the interface: whereas the electron is located at the TiO2 conduction band or traps, the hole is an adsorbed radical formed upon the electron transfer from the molecule to the semiconductor. If in solution there are scavengers for electrons and holes, recombination would be prevented and a photochemical reaction would be induced by the Raman laser probe. This should be reflected in series of successive Raman spectra. In fact, RRS for both salicylic and ascorbic acids evolves with time during continuous laser illumination. It is remarkable that the changes are significantly faster when working with nanoporous thin films than for slurries.
Fig. 5 shows the initial spectrum obtained for salicylic acid adsorbed at a TiO2 nanoporous thin film together with successive spectra taken at different times. As observed, after a few seconds the spectrum is fully dominated by an intense fluorescence, probably coming from a product formed at the nanoporous film. In addition the only remaining bands are very broad and linked mainly to ring vibrations in the condensate. The anodic polymerization of salicylic acid has been shown to occur on various electrodes (on glassy carbon, Pt, etc.) in acetonitrile solution [41,42]. Photoanodic polymerization also seems occurs on TiO2 electrodes [41]. More work is needed to identify the products giving fluorescence.
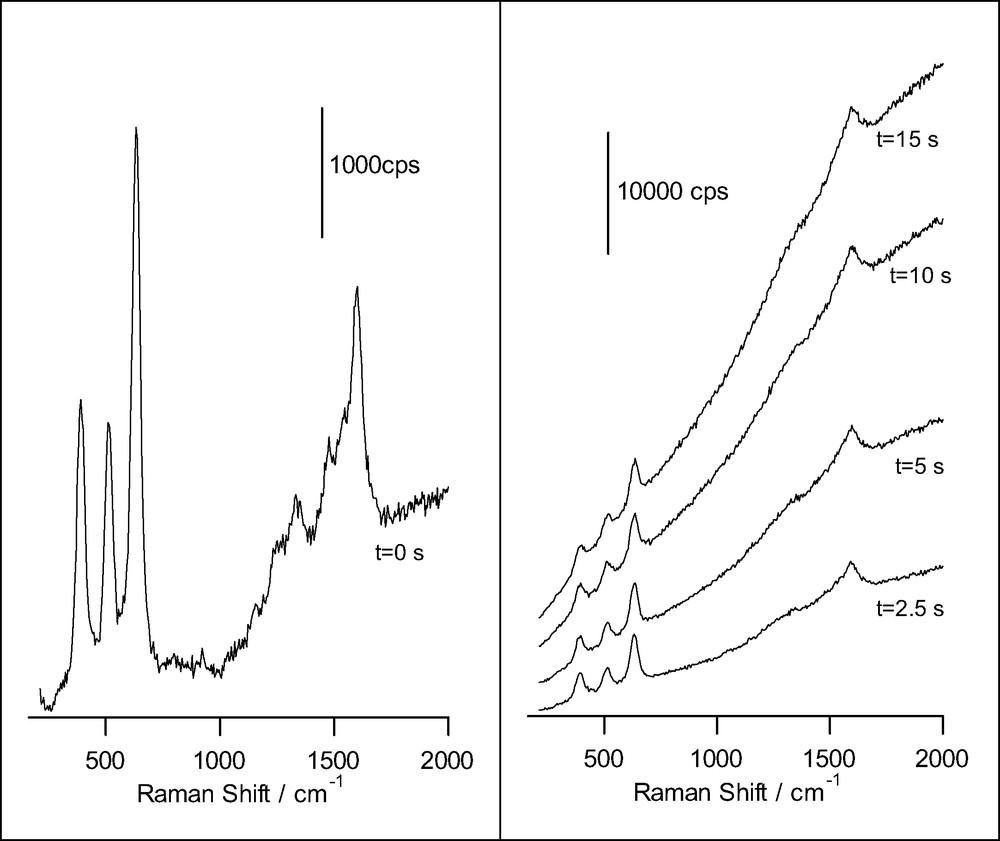
Raman spectra obtained for a nanoporous film of anatase on FTO conducting glass, in presence of 14.5 mM salicylic acid and 0.1 M HClO4 (acquisition time: 0.5 s), for different illumination times. The excitation line was provided by an Ar+ laser.
A different behavior is found for ascorbic acid adsorbed at anatase nanoporous thin films (Fig. 6). In this case, the evolution during illumination consists simply in the attenuation of the spectral features, which points to the fact that in this case the photooxidation product desorbs from the surface. No fluorescence is developed in this case, suggesting that condensation does not occur. Our observations are in agreement with proposals made on the basis of photoelectrochemical measurements suggesting that the photooxidation of ascorbic acid produces dehydroascorbic acid, which does not adsorb [28].
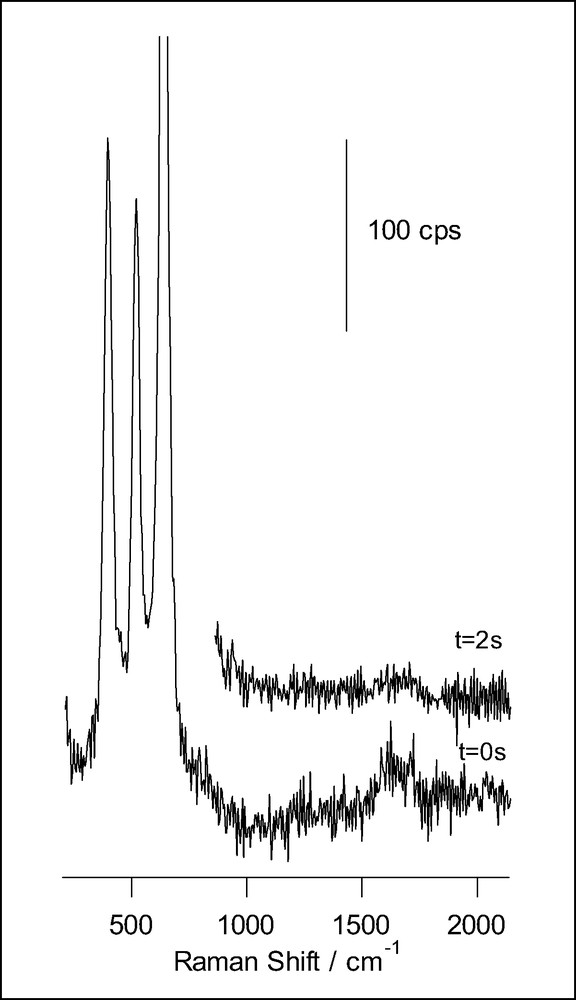
Raman spectra obtained for a nanoporous film of anatase on FTO conducting glass, in presence of 14.5 mM ascorbic acid and 0.1 M HClO4 (acquisition time: 0.2 s), for different illumination times. The excitation line was provided by an Ar+ laser.
We have presented time-dependent spectral results for the heterogeneous photooxidation of two TiO2 surface modifiers, which stress the utility of Raman spectroscopy not only for elucidating the adsorption geometry of species, but also to obtain in situ information on the fate of the charge transfer complex once excited. More work along these lines is being developed in our laboratory.
4 Conclusions
In this paper, we have tried to illustrate how Raman spectroscopy can be used in adsorption studies at TiO2 slurries or nanoporous films by taking advantage of two different enhancement mechanisms: surface enhancement by plasmonic excitation and resonance.
As for SERS, we have deposited a thin film of anatase nanoparticles onto a roughened gold electrode and studied the adsorption of typical organics such as methanol, formic acid or phthalic acid [22], all of them dissolved in electrolytic solutions. Due to the fact that nanostructured thin films possess a high inner surface area, a significant amount of species adsorbed on TiO2 are in the spatial region where electromagnetic intensification (through plasmonic excitation of the Au substrate) is strong enough for allowing detection of adsorbates. Our approach is therefore well suited for the study of the adsorptive behavior of nanoporous semiconductor thin films (electrodes), which are being increasingly employed in fundamental and applied research in photocatalysis and photoelectrochemistry. This technique of Raman enhancement has the advantage of its lack of molecular specificity, being thus applicable to most adsorbates, without the restriction of needing resonant effects. One difficulty sometimes appearing in our experimental procedure is linked to the fact that adsorption on the gold substrate may completely obscure signals coming from adsorbates on anatase, as in the case of phthalic acid. This has been overcome applying a positive potential to the gold substrate in such a way that its surface becomes oxidized. Blank experiments (with the bare gold substrate) indicate that when the gold surface is oxidized, the adsorption of most organics is inhibited.
It is noteworthy that these adsorption studies provide additional evidence to kinetic modeling of the behavior of illuminated TiO2. In particular the adsorption of formic acid (as formate) and the apparent lack of methanol adsorption in aqueous solutions supports a recent kinetic study aiming at the discrimination between direct (via free valence band holes) and indirect (via OH radicals) mechanisms in heterogeneously catalyzed photooxidations [23].
We have also extended RRS to the study of adsorbates typical of photocatalytic studies for the TiO2/solution interface. Concretely, spectra corresponding to adsorbed salicylic and ascorbic acids on anatase nanoparticulate films or slurries are presented. These organics have the advantage of showing resonance only in the adsorbed state because of the formation of ligand-to-metal charge transfer complexes, which avoids interference from solution species. In addition to furnishing vibrational information on the adsorbed species, Raman experiments provide some information on the products resulting from the oxidation of the adsorbed species induced by the visible laser light.
Acknowledgements
T.L.V. is grateful to the MECD (Spain) for the award of a FPU grant. The Spanish DGI (MCyT) is acknowledged for financial support through project No. BQU2003-03737. We also thank the S.S.T.T.II. and the Vice-Presidency of Research of the University of Alicante.