

1 Introduction
Heterogeneous photocatalysis, using titanium dioxide as the photocatalyst of choice, is a useful technique for the degradation of many contaminants in air, in water and on solid surfaces [1]. The general scheme for the photocatalytic destruction of organic compounds involves the excitation of this semiconductor by irradiation with photons of supra-bandgap energy and migration of the electron-hole pairs to the surface of the photocatalyst, where the holes may be trapped by H2O or OH– adsorbed on the surface, thus forming highly reactive hydroxyl radicals. Since oxidation is governed by a free radical mechanism, it is expected to operate in a manner that hardly differentiates between different contaminants, provided that these molecules adsorb on the photocatalyst's surface. [2] Indeed, competitive adsorption is often the key in the photodegradation competition as observed by Zahraa et al. who studied the co-degradation of atrazine and salicylic acid [3].
The lack of sensitivity to the type of contaminants seems to be benevolent at first glance, since it means that there is no need to pre-design the photocatalyst for a certain contaminant. It suggests that this photocatalyst may handle air/water streams containing mixed contaminants, without much need for on-line characterization. Nevertheless, a second thought alters this picture. Mixed streams may contain hazardous contaminants together with organic contaminants of low toxicity. In many cases, the toxic stuff is in low concentration, whereas the less toxic is the majority. It is obvious that in such cases, it is preferable to degrade the more toxic stuff, even at the expense of lower degradation rates for the less harmful components. Moreover, many low-toxicity contaminants can be degraded by biological means, whereas many of the highly hazardous materials are non-biodegradable, due to their high toxicity. The fact that the adsorption coefficient of many of the chlorinated xenobiotic compounds on titanium dioxide is very low, due to their hydrophobic nature, aggravates this problem.
The problem of mixed streams is often coupled with the formation of toxic intermediates, and the need to prevent their emission. Here, two good examples are the photocatalytic degradation of trichloroethylene (TCE) in the gas-phase, which produces phosgene and dichloroacetyl chloride [4], and the photodegradation of the pesticide diuron [5]. A synergetic combination of several techniques, for example photocatalysis and biological treatment, may provide the right solution. That way, combining sonolysis and photocatalysis was suggested as a means to break down chlorinated aromatic compounds; the former technique breaks down non-polar materials into more polar compounds, while the latter method is superb in mineralizing polar molecules [6].
Alternatively, or even in parallel, it could be most beneficial to design ways, by which the degradation of a specific contaminant, in a stream containing several compounds, is preferred. Unfortunately, studies where the main aim is obtaining preferential photocatalytic degradation in mixed streams are exceptionally rare at present. In many cases they represent an existing need to solve a very specific problem (for example, the work of Ireland et al., who studied the degradation of a mixture containing 16 polyaromatic hydrocarbons, extracted from motor oil contaminated soil [7]) and are less aimed at attacking this issue on more generalized grounds.
Yet, a lot of useful knowledge can be obtained by screening cases where relative photodegradation rates were altered as a consequence of changing operational parameters, or, otherwise, following tailoring of the photocatalyst. Generally speaking, most of these parameters act through controlling the adsorption of specific molecules onto the photocatalyst surface. These parameters, grouped into process parameters and photocatalyst-related parameters, are described herein.
2 Process parameters that affect preferential photodegradation
This section describes studies on the effect of various operational parameters on changing the photodegradation rate ratio between two or more contaminants. It includes those process variables that can be changed while operating with a given, un-modified, titanium dioxide photocatalyst. Such variables are, for example, the temperature, pH, presence of ions, or even UV wavelength. It is noteworthy, that in light of the scarce data existing in the literature on the photodegradation of mixed contaminants, part of this survey is based on comparing data on the degradation rates of two (or more) single-contaminant streams that were treated under exactly the same conditions. The validity of this comparison, which neglects cross-influence is discussed in Section 4.
2.1 Temperature and residence time
One of the first works that tackled the issue of preferential photodegradation of a specific contamimant in a stream containing several compounds is that of Muradov et al. [8], who studied the photodegradation of airborne nitroglycerine in the presence of ethanol and acetone. Their aim was to facilitate the recovery of the solvents, while degrading emitted nitroglycerine vapors. For that, various process parameters were tested, among which were the temperature and residence time. Most of their experiments were performed at elevated temperatures, between 80 and 200 °C, where thermocatalysis takes place in parallel to photocatalysis. In the absence of light, thermodegradation rates followed the adsorpticity of the components, i.e. NG > ethanol > acetone. This order was slightly different in photocatalytic measurements with P25 TiO2 that were performed at 80 °C, where the photodegradation rate of ethanol was slightly higher than that of NG, but by far larger than that of acetone. As expected, the shorter the residence time was, the better the prefertiability was (at the expense of lower conversion). Another difference between the thermocatalytic process and the photothermocatalytic process was manifested through the effect of temperature. In the dark, the highest degradation rates ratio between NG and ethanol/acetone was obtained at the lowest temperature, whereas in the photocatalytic oxidation, high temperatures were favorable. These findings correlate to some extent with a notion that, for this system, the importance of adsorption as the dominant mechanism is higher in thermocatalysis than in photothermocatalysis.
2.2 pH
The reported isoelectric point (point of zero charge) for TiO2 varies to some extend between pH 6 [9,10] and pH 7.5 [11,12]. This means that pH values higher than that are favorable for the adsorption of positively charged contaminants, while pH values lower than the PZC are favorable for the adsorption of negatively charged contaminants. Hence, controlling the pH of the medium, thus controlling the surface charge, may serve, at least potentially, as a mean to obtain preferential degradation [13,14]. In this context, a straight-forward example is the pH effect on the photocatalytic reduction of Hg(II), in a mixture prepared by dissolving HgCl2 and K2Cr2O7 [15].
It should be emphasized that the pH effect is a complex one, as the pH affects not only the adsorption of charged contaminants, but also alters the concentration of surface hydroxyls and shifts the position of the valence and the conduction bands. A decrease in pH of one unit results in a shift of +59 mV in the position of the bands, as reflected, in the absence of any contaminants, by a pH dependence of the photocurrent [14].
Although the pH primarily affects the adsorption of charged contaminants, it has also a role on the photocatalysis of those neutral molecules that tend to dissociate into charged species. One may need to take into consideration that in that case, the pH affects not only the surface of the photocatalyst, but also the dissociation of the contaminant. A relevant example is the codegradation of acetic acid and 2-chlorobiphenyl [16]. Here, the photodegradation rate of acetic acid peaked at a pH of 4.6, in accordance with its pKa (4.7), reflecting the balance between surface charging and contaminant charging. In contrast, the degradation of the neutral 2-chlorobiphenyl, was not affected by changing the pH. Similarly, the photodegradation rate of formic acid (pKa = 3.75), peaked at a pH of 3.4 [17]. Recently, this approach was tested deliberately in order to obtain preferential degradation in the photodegradation of a mixture of 4-hydroxybenzoic acid and benzamide [18]. As expected, at pH 4 the degradation rate of 4-hydroxybenzoic acid was much higher than that of benzamide, whereas at pH 8, the opposite was observed.
In certain cases, the photodegradation of neutral molecules leads to the formation of charged species. If this happens, the pH will affect mainly the degradation of the intermediates. Hence, it could be expected to observe a pH effect on the mineralization rate, but not on the photodegradation of the neutral reactants, except for a minor effect due to competitive adsorption. Indeed, while the photodegradation of 4-chlorophenol was hardly influenced by the pH, its overall mineralization rate (measured in terms of total organic carbon reduction) changed considerably, decreasing strongly at pH > 7 [19].
The pH effect seems to operate in several more ways. One of these is its effect on the reaction between protons and superoxide ions to form OOH radicals:•O2– + H+ ↔ •HO2 pKa = 4.88.
The OOH may contribute to the formation of H2O2, and thus to the production of extra hydroxyl radicals. Indeed, elevating the pH from 3 to 11 had a detrimental effect on the photodegradation of 2-chlorophenol (from k = 0.02 min−1 to k = 0.006 min−1) that could not be rationalized only on the basis of surface charge, but rather by a combined mechanism that involves surface charge and OH production effect [20].
Overall, although performing the photocatalytic process at a pre-determined pH helps to differentiate between positively charged contaminants and negatively charged contaminants, the pH effect is not sufficient as a means to obtain high reactant selectivity due to its lack of specificity, its mild effect, its interrelation with other factors and the fact that most contaminants are neutral.
2.3 Presence of ions in the feed
The effect of common inorganic anions on the photocatalytic degradation of salicylic acid, aniline, and ethanol was studied by Abdullah et al. [21]. It was found that the presence of chlorides, sulfates and phosphates reduces the oxidation rates of these compounds, nevertheless, the extent by which the rates were reduced, at a pre-determined pH (4.1), was different for each compound. For example, the presence of 0.001 M Na3PO4 reduced the photodegradation rate of salicylic acid by 64% (from 85 to 30 μg carbon min−1) but reduced that of ethanol by no more than 44% (from 41 to 23 μg carbon min−1). This difference apparently suggests a way for preferential degradation. Nevertheless, as shown above, the effect of the co-solutes on the selectivity is very mild, and cannot be regarded as practical. To this one should add the fact that in all cases the photodegradation rates in the presence of the cosolutes were lower than in the absence of co-solutes, a feature that is rarely desired.
An interesting way to separate between Chromium(VI) ions and Hg(II) ions was presented by Wang et al. [22]. The method is based upon the complexation of Hg(II) following the addition chloride ions. This complexation retards the photocatyalytic reduction of Hg(II) without affecting that of Cr(VI), thus facilitating to separate the two, despite their similar redox potential (0.85 V for Hg2+/Hg and 0.82 V for Cr2O72–/Cr3+).
2.4 Type of solvent
In aqueous solutions, adsorption might be modified by solvents in a manner that is specific for each type of contaminant. In that sense, the use of solvents may alter the ratio of photodegradation rates between various contaminants. To the best of our knowledge, this effect was never considered as a mean to obtain preferential degradation. Nevertheless, it was well demonstrated in the work of Almquist and Biswas [23], where the adsorption of cyclohexanol and cyclohexanone on TiO2 in various liquid phase solvents such as cyclohexane, n-hexane, isopropanol, acetone, benzene, and chloroform was measured. It was shown that the ratio of alcohol to ketone adsorption on TiO2 could vary by a factor as large as 22 by using appropriate solvents. Accordingly, one may claim that there is ground to believe that the type of solvent may provide another route towards preferential degradation. Alas, the type of solvent, and in particular organic solvent, is not usually considered as a parameter of choice (in particular since the solvents themselves tend to photocatalytically degrade).
2.5 Irradiation with sub-bandgap light
The formation of charge transfer complexes between adsorbed contaminants and the titanium dioxide surface may affect reactivity and product formation. In certain cases, for example in the case of 2,4,5-trichlorophenol, the formation of the complex might depend on irradiation with sub-bandgap light [24]. Hence, in principle, such sub-bandgap irradiation at a specific wavelength may assist in promoting the supra-bandgap photodegradation of specific molecules. To date, we are not aware of any attempt to utilize this effect for this purpose. Still, such route should not be ruled out.
2.6 Photocatalyst concentration
For liquid phase treatment, the concentration of the photocatalyst can be regarded as an operational parameter. It is obvious that changing the photocatalyst concentration, with its impact on the total surface area, the average contaminant–photocatalyst distance, the penetration depth of the UV light, etc., affects both the photodegradation rates and mineralization rates. This effect can vary from one contaminant to the other, in a way that may suggest using a certain concentration in order to achieve the optimal ratio between the degradation rates of several co-existing contaminants. As an example, one may outline the mineralization and degradation of 1,2-diphenylhydrazine versus that of benzidine [25]. Here, at the range of concentrations that were studied (0.5–5 g l−1), the rates for the former were found to increase as a function of concentration, whereas the rates for the latter peaked at a catalyst concentration of 2 g l−1. Within this concentration range, the ratio between the photonic efficiency for the mineralization of DPH to that of benzidine changed from 3.2 at a concentration of 0.5 g l−1, to 2.8 at a concentration of 2 g l−1 and back to 4.1 at a concentration of 5 g l−1.
3 Photocatalyst-related parameters
The existing of a variety of methods to produce titanium dioxide, and the ability to modify the size, the crystalline phase, and the surface of the photocataylst provide more ways for obtaining preferential photodegradation. Moreover, it enables to pre-design photocatalysts with very high specificity, as will be described in the following section.
3.1 Un-treated TiO2 made by different sources
Degussa's P25, with its 80/20% anatase/rutile titanium dioxide, is often considered as the benchmark for TiO2 photocatalysis, due to its superb activity. Nevertheless, there are contaminants, for which other commercially available TiO2 are much more suitable. In order to demonstrate the influence of the type of photocatalyst on preferential degradation, we have calculated the ratio between the photonic efficiencies measured by Muneer et al. [25] in the degradation of 1,2-diphenylhydrazine, to those measured by them in the degradation of bezidine. These ratios, for various types of TiO2 particles are presented in Fig. 1.
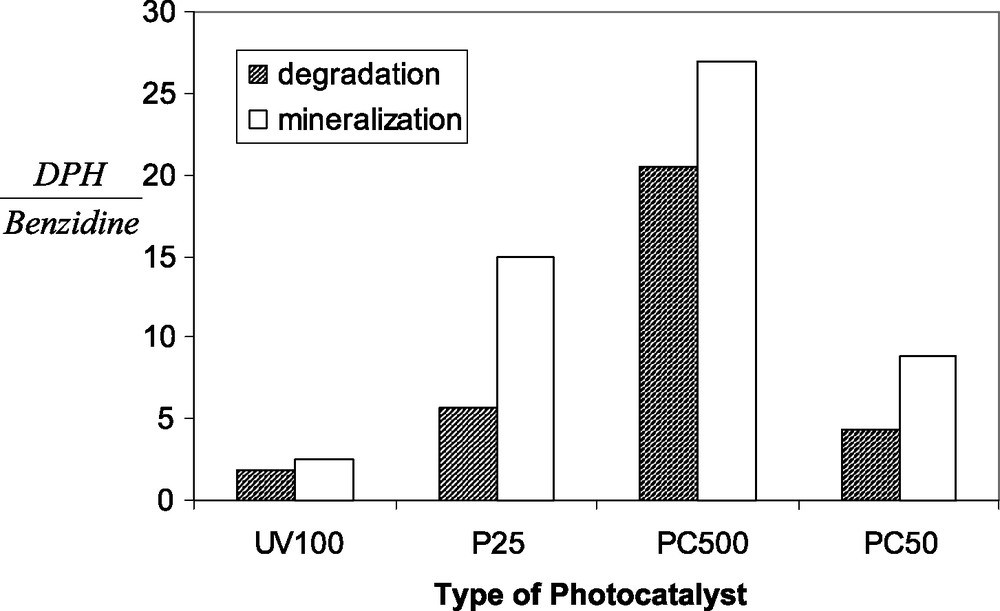
The ratio between the photodegradation and mineralization rates of 1,2-diphenylhydrazine (DPH) to that of benzidine for various types of titanium dioxide (based on Ref. [25]).
As presented in the figure, these ratios change quite considerably between different types of titanium dioxide particles; replacing Hombikat UV100 with P25 particles changes this ratio by a factor of 3, and likewise, this ratio may change by a factor of 10 (!) simply by using PC500 particles (Millennium Inorganic Chemicals) instead of UV100. A possible reasoning, suggested by Muneer et al. to explain the difference between UV100 and P25, is based on Martin's claim that the high activity of P25 is due to slow recombination rate, whereas the high activity in the UV100 is due to fast interfacial electron-transfer rate [26]. In that case, P25 is adequate for the degradation of molecules that their adsorption (and desorption of intermediates) is slow, whereas UV100 is adequate for molecules that their adsorption (and desorption of intermediates) is relatively fast. Another factor related to the difference between P25 and Hombikat UV100 is the point of zero charge, claimed to be located at a pH 6.2 for Hombikat UV100 and at pH 7.1 for Degussa P25, thus affecting the adsorption of Hg2+ during its photocatalytic reduction [15].
Blount et al. [27] have reported on the preparation of a transparent film made by a sol–gel method. The phocatalytic activity was evaluated based on the rate of CO2 formation during the photodegradation of acetaldehyde and acetic acid. Analyzing their results reveals that with the sol–gel film, the rate of CO2 production during acetaldehyde degradation was 2.5 times higher than the rate of CO2 production during the acetic acid degradation. In contrast, this ratio was no more than 1.17 when P25 was used. A correlation with the adsorption isotherm of the contaminants, where the ratio between the specific adsorption of acetaldehyde to that of acetic acid was slightly higher on the sol–gel film than on P25, was found. No reasons for the preferential adsorption of acetaldehyde were given.
Some preferential degradation was reported in the case of sulfated TiO2, i.e. TiO2 that was prepared by reacting H2SO4 with Ti(OH)4. Here, it was found that the ratio between the mineralization rate constant of heptane to that of toluene was 5.75 for the sulfated titanium dioxide, while for non-sulfated TiO2, made by calcining unsulfated Ti(OH)4, this ratio was no more than 2.7. This selective enhancement in the degradation of heptane, correlated well with increased coverage and adsorption strength on the strong Lewis acid sites produced by sulfation [28].
3.2 Doping
It is well established that doping by noble metals, such as Pt or Pd may accelerate the cathodic process of oxygen reduction [29]. This can be used in order to differentiate between those contaminants that differ significantly in their photodegradation mechanism. For example, metallization increases the degradation rate of methanol and ethanol that degrade through oxidation, but reduces the degradation rate of trichloroethylene, chloroform, and dichloropropionic acid (DCP) [30]. Likewise, platinization of Hombikat UV100 resulted in twofold increase in the photodegradation ratio between dicholoacetic acid (DCA) and 4-chlorophenol [31]. Reduction in the photodegradation rate of 4-CP following platinization was observed also by Al-Sayyed et al. [32], in contrast to the known enhancement for many contaminants. The various parameters that affect the extent of rate modification upon doping with Pt or Pd are described by Lee and Mills [33], who reported enhancement factors over a wide range (0.1–8).
The effect of metal ion dopants on the photocatalytic reactivity of quantum-sized TiO2 particles was studied by Choi et al. [34], who made a correlation plot between the oxidation (chloroform degradation) quantum yield and the reduction (dechlorination of carbon tetrachloride) quantum yield, showing that doping with V(IV) increases reduction relative to oxidation, while doping with Ru(III) increases oxidation relative to reduction.
Silico-tungstic acid (STA = H4SiW12O40·n H2O) is known as a multi-electron redox agent, which can interact with photogenerated electrons and holes. It was found that doping with STA completely hindered the degradation of ethanol and acetone but had no effect on the photocatalyst activity towards the destruction of nitroglycerin. In that manner, complete selectivity can be achieved, albeit at the expense of reducing the overall photocatalytic activity [8].
Doping with lanthanide oxide was found to increase the photodegradation rate of p-chlorophenoxyacetic acid [35]. It was claimed that this increase was due to the formation of a complex between the lanthanide ion and the substrate. Although specificity towards other contaminants was not analyzed, it is conceivable that this enhancement is not specific, since lanthanides are known for their ability to form complexes with acids, amines, aldehydes, alcohols and thiols. The specific interaction between the dopant and the acid affected intermediates distribution, as the only intermediate that was found was p-chlorophenol, instead of hydroquinone and chloroquinone that are found in the case of pure TiO2. Another example is the loading of nanoclusters of Ag that was found to have a net positive effect on the photocatalytic reduction of bis(2-dipyridyl)disulfide (RSSR) to 2-mercatopyridine, due to enhanced adsorption of RSSR on the silver nano-domains [36].
3.3 Overcoating titanium dioxide
Overcoating the photocatalyst with a material that has specific affinity towards a certain contaminant (or a group of contaminants) can serve, in principle, as a way to photodegrade selected contaminants. In most cases where this method was used, the overcoat consisted of molecules having hydrophobic moieties, thus increasing the adsorption, and hence the degradation, of hydrophobic contaminants. That way, grafting of n-octyltriethoxysilane onto TiO2 surface was found to be very useful for the degradation of the endocrine disrupter 4-nonylphenol in the presence of concentrated phenol [37]. Likewise, the degradation rate of the dye Rhodamine B, which does not adsorb appreciably on pure TiO2, was increased following hydrophobic modification with methytrimethoxysilane [38]. In that manner, titanium dioxide particles partially coated with a hydrophobic organo-silicone layer [39] were found to be very efficient in the photocatalytic destruction of Permatrin [40], a water insoluble pesticide.
While this method may enhance the photodegradation of hydrophobic contaminants, it suffers heavily from the fact that the grafted molecules themselves also degrade, as was found before by Fujishima's group, who used such degradation as a means for fast evaluation of the photoactivity of different types of titanium dioxide (the so-called ‘photosinking phenomena’ ) [41]. One way to prevent degradation of adsorptive sites is to coat anatase TiO2 with a thin layer of inorganic carbon, produced by thermal carbonization of poly(vinyl alcohol) under N2 environment [42]. This layer was found to be very beneficial in promoting the adsorption of methylene blue, and, as a consequence, in promoting its photodegradation.
Another example for the effect of overcoating is the modification of titanium dioxide nanoparticles with the electron-donating chelating agent arginine that binds to TiO2 through its COO– functional group [43]. This modification resulted in enhanced specific adsorption of nitrobenzene, due to interaction of the π-system of the nitrobenzene ring with the arginine amino group, which led to faster reductive photodecomposition, compared with un-modified TiO2.
3.4 Adsorption on inert inorganic domains located in the vicinity of active sites, followed by diffusion: the “inorganic adsorb-and-shuttle” approach
The ‘inorganic adsorb-and-shuttle’ method is similar to the one presented in the previous section. The basic concept is the adsorption of the contaminants on inorganic domains located at the vicinity of the photocatalyst, followed by their surface diffusion from the inert adsorptive sites to the photocatalytic sites. The net result is an increase in the concentration of the contaminants at the photodegradation sites. That way, one may induce not only enhancement of mass transport, based on the improved qualities of the adsorptive sites, but actually can (at least partially) ‘map’ any adsorption specificity that the inert adsorptive sites have, into preferential photodegradation.
Mixing activated carbon and titanium dioxide was found as a way to increase the photodegradation rate, as was demonstrated in the degradation of phenol [44]. The fact that this synergy effect varies from one pollutant to another (for example: a synergy factor of 2.4 for 4-chlorophenol, but only 1.3 for 2,4-dichlorophenoxyacetic acid) [45] may provide a means to enhance the degradation of certain contaminants, in particular if one takes into consideration that the specific synergetic effect varies also with the type of activated carbon that is used (i.e. hydrophobic made by high temperature activation, versus hydrophilic, made by low temperature activation) [45]. It should be noted that the distance between the adsorptive sites and the photocatalytic sites is highly important. Construction of composite particles made of activated carbon and titanium dioxide increased considerably the photodegradation rate of Rhodamine-6G (R6G), while simple mixing reduced it, due to competitive adsorption [46]. The adsorptive substrate also has an effect on the type of emitted products; for example, the use of composite particles made of P25 and activated carbon totally prevented the appearance of any intermediates in the photodegradation of gas-phase TCE [47].
A thorough work, comparing the effect of various adsorptive substrates such as silica, alumina, activated carbon and zeolites, on the photodegradation of propyzamide [48], propioaldehyde [49], and pyridine [50], was performed by the group of Yoneyama, who even calculated the diffusion coefficient of adsorbed propionaldehyde on the adsorptive substrates [51]. It was established that in cases where the adsorption constant on the adsorptive sites is low, the decomposition rate is determined by the amount of adsorbed contaminant, while if the adsorption constant is very high, plenty of adsorbed molecules can be found on the support, yet, the degradation rates are low, due to insufficient mobility.
Although the inorganic ‘adsorb-and-shuttle’ may significantly increase the photodegradation of contaminants that hardly adsorb on titanium dioxide, and may help in preventing the emission of intermediates, its specificity is inherently quite low, as it operates on a class of contaminants, and since the ability to ‘tailor’ the adsorptive substrate is limited.
3.5 Adsorption on inert organic domains located in the vicinity of active sites, followed by diffusion: the ‘organic adsorb-and-shuttle’ approach
Recently, it was suggested [52] to introduce immobile organic molecular recognition sites (MRS) on inert domains, located on or in the vicinity of the photocatalyst. These pre-designed molecular recognition sites would physisorb selectively target molecules, that would surface-diffuse from site to site towards the interface between the inert domains and the photocatalytic domains, where they would be destroyed (Fig. 2).
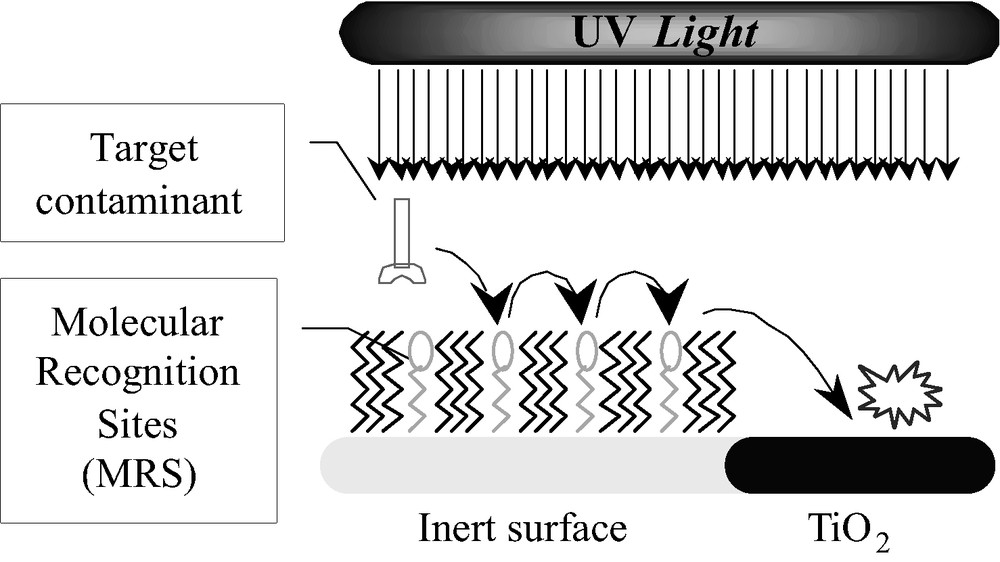
The ‘specific adsor-and-shuttle’ concept.
Since the molecular recognition sites are organic, care has to be taken to prevent their destruction by the hydroxyl radicals that are formed on the titanium dioxide surface. Obviously, this means that the MRS should not be constructed directly on the photocatalytic titanium dioxide surface. Moreover, it is needed to assure that MRS located on inert sites, adjacent to photocatalytic domains are not prone to an attack by oxidizing species, since such oxidizing species can destroy organic molecules anchored on oxide surfaces as far as 40 µm away from the photocatalytic domains [53]. This instability towards remote degradation was not found when self-assembled monolayers anchored through a thiol functional group to metallic stripes were examined [54].
The first system demonstrating this approach used β-cyclodextrin as the molecular recognition host, and 2-methyl-1,4-naphtoquinone (2MNQ) as the model contaminant [55]. The host molecules were chemisorbed on micrometer-sized gold stripes located next to microns-sized titanium dioxide stripes on silicon wafers. β-Cyclodextrin is a cyclic oligosaccharide, made of seven glucopyranose units forming a torus-like structure with a cavity of 0.78 nm in diameter. This molecule was chosen to demonstrate the ‘A-and-D’ approach based on the well-known size-selective affinity between this molecule and a variety of apolar guests whose size matches that of the cyclodextrin cavity. Among these guest molecules are drugs, food flavors, pesticides, herbicides, and dye-stuffs [56].
Fig. 3 presents the kinetics of photocatalytic degradation of 2MNQ physisorbed on the thiolated cyclodextrin, as deduced from the reduction in the area of its 1666 cm−1 IR peak, for structures having adsoptive domains of different sizes. As shown in the figure, the smaller the domain width was, the faster the photodegradation of the 2-methyl-1,4-naphtoquinone was. For example, 20% degradation was obtained with the 5 μm width structures after 5 min, whereas the same extent of degradation with the 40 μm width structures required 90 min. This effect of the domain size is in line with the notion that the 2MNQ molecules were adsorbed on the molecular recognition sites, from where they had to diffuse to the photocatalytic titanium dioxide domains. That the degradation was due to 2MNQ diffusion from the MRS to the TiO2 domains and not due to OH diffusion is supported by the observation that the degradation of the 2MNQ was not accompanied by the degradation of the molecular recognition sites. A control experiment assured that the degradation was not due to direct photolysis, or to the presence of gold per se.
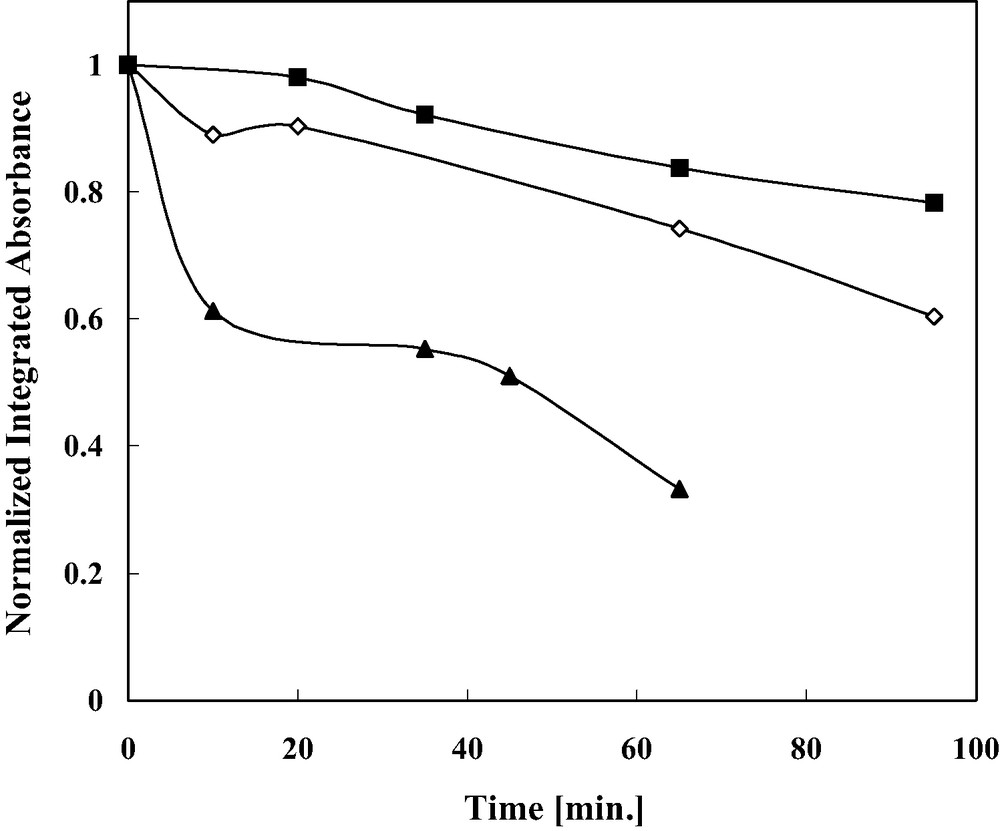
The photodegradation kinetics of 2-methylnaphtoquinone on substrates containing TiO2/Au stripes of different width, onto which thiolated cyclodextrin was chemisorbed. In all samples, the width of the gold stripes was equal to that of the TiO2 stripes. Filled triangles represent stripes of 5 μm in width, empty diamonds represent stripes of 20 μm in width, and filled squares represent stripes of 40 μm in width.
A comparative study on the ability of these structures to enhance the degradation of selected contaminants was done in the liquid phase by comparing between the rate of disappearance of 2MNQ vs that of benzene. Table 1 presents the extent of decrease in the concentration of both contaminants following 30 min of exposure, and the ratio between the relative degradation of 2MNQ to that of benzene, for solutions containing one contaminant at a time and for solutions containing both contaminants at the same time. A comparison between the three types of wafers reveals that for both types of solutions the MRS-containing wafers were significantly more efficient in photodegrading 2MNQ than wafers that did not have the MRS (60% degradation vs. 18.7% degradation, i.e. more than 200% faster). In contrast, the rate of benzene photodegradation with the cyclodextrinated wafers was lower than the rate on wafers that did not have the MRS. The overall effect is reflected in the ratio between the degradation rate of 2MNQ to that of benzene. Here, an increase of more than 10 times (8.1:0.75) was found upon comparing solutions containing one contaminant at a time, and an increase of six times (4.2:0.71) was found upon comparing solutions containing a mixture of the two contaminants. Hence, the feasibility of enhancing the preferential degradation of selected contaminants by constructing molecular recognition sites at the vicinity of titanium dioxide domains was clearly demonstrated.
The extent of decrease in the concentration of benzene and of 2-methylnaphtoquinone, following exposure of vessels containing the various types of wafers to 365-nm light, and the ratio between the extent of degradation of 2MNQ to that of benzene
| Solutions containing one contaminant | Solutions containing both contaminants | |||||
| TiO2 | Au/TiO2 | TCD/Au/TiO2 | TiO2 | Au/TiO2 | TCD/Au/TiO2 | |
| 2MNQ | 22.3% | 18.7% | 60% | 13% | 19.8% | 32.6% |
| Benzene | 26% | 25% | 7.4% | 26.3% | 28% | 7.7% |
| 2MNQ/Benzene | 0.86 | 0.75 | 8.11 | 0.49 | 0.71 | 4.23 |
Both 2MNQ and benzene are known to form inclusion complexes with β-cyclodextrin [57,58]. The negative effect measured with benzene can be explained by very low surface mobility of benzene, probably due to high complexing energy between the host and the guest. Such an explanation goes well with the notion that benzene penetrates very deep into the cavity of β-cyclodextrin, forming a very tight 1:1 complex [59].
Another model contaminant that shows enhanced rate of photodegradation due to the presence of β-cyclodextrin close to titanium dioxide microdomains is the dye-stuff Chicago Blue Sky 6 (CB). Here, an aqueous solution containing a mixture of 0.01 mM of CB and 0.01 mM of the dye-stuff R6G was exposed to 365 nm UV light (0.09 mW cm–2). The degradation of the CB dye and that of R6G was calculated based on changes in their UV–VIS spectra, as reflected in their 625 and 527 nm peaks, respectively. It was found that the degradation of the CB was 1.7 times (70%) faster when using the MRS-containing samples. This enhancement was not observed for R6G. It was suggested, albeit not proved, that the enhancement in the photodegradation of Chicago Blue is closely related with its structure.
Recently, a new ‘adsorb-and-shuttle’ (A and S) system, based on 1,1-mercaptoundecanoic acid (MUA), whose proton was substituted by Cu2+ (MUACu), was presented [60]. This system was designed for the photodegradation of diisopropyl methylphosphonate (DIMP), known as a nerve agent (sarin) simulant. Similar to the previous case, the measurements were performed on silicon wafers coated with alternating microstripes of photocatalytic titanium dioxide and inert gold substrates onto which the MRS was chemisorbed. More details on this study are presented elsewhere [61].
Fig. 4 presents the evolution of the IR spectrum during a typical photocatalytic experiment, done in a closed vessel, with MRS-coated patterned substrates. Typical DIMP peaks are observed at 917, 990, 1108, 1263 and 2982 cm−1. The evolution of the spectrum upon photodegradation is clearly demonstrated in traces b, c, d and e. These changes correlate well with the degradation of DIMP, the formation of acetone (1735, 1367, 1212 cm−1) as an intermediate product, and the formation of CO2 (2348 cm−1) and water as the final products. A significant growth in CO (2178, 2119 cm−1) concentration is noticed after 30 h, probably due to some shortage of oxygen in the closed vessel. The evolution of the spectra in experiments with patterned substrates that had not previously coated with MUACu was qualitatively similar. Control experiments, done in the absence of TiO2 substrates showed no evolution of the DIMP spectrum upon illumination, indicating that the degradation of DIMP was due to photocatalysis and not due to direct photochemistry.
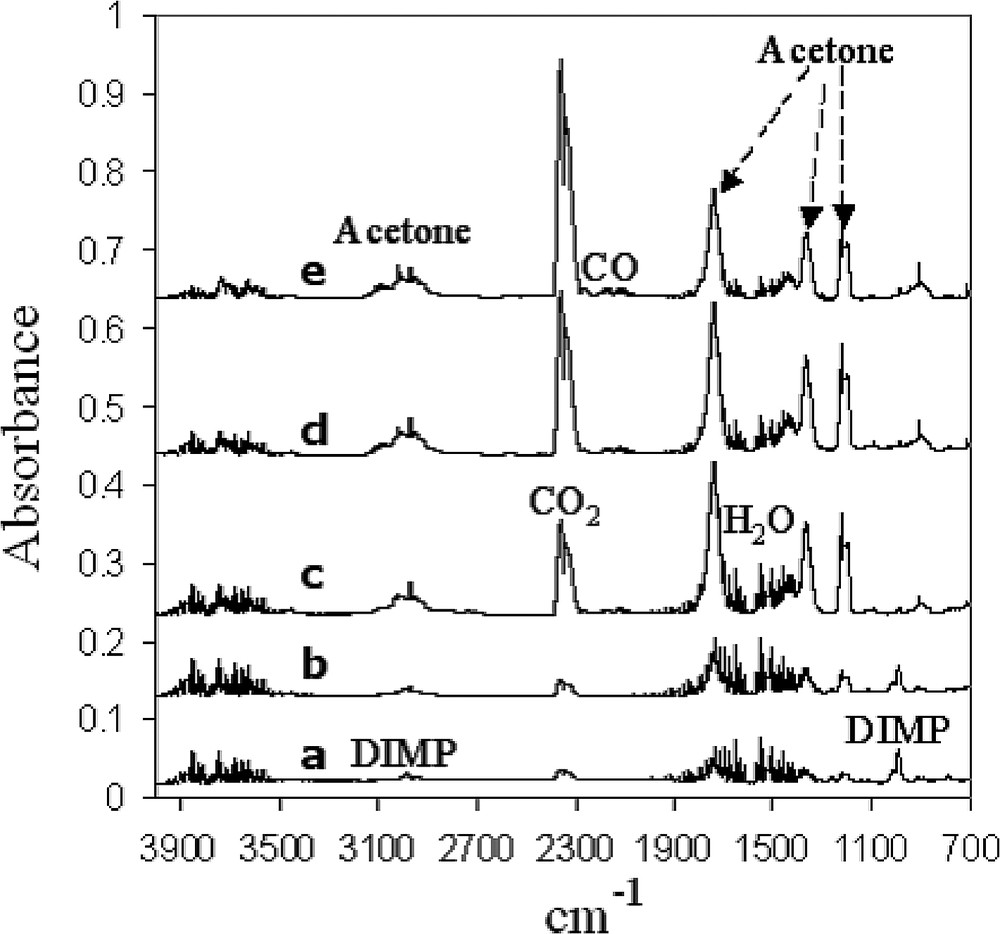
The evolution of an FTIR spectrum upon photodegradation of DIMP on TiO2/MUACu on Au: a-before UV exposure, b–e-following UV exposure for 3, 19, 39, 60 h, respectively.
Based on the evolution of the spectra it can be deduced that the photocatlytic degradation of DIMP proceeds as describes in Fig. 5, i.e. through the degradation of DIMP to the intermediate product acetone, later to be decomposed. This process occurs simultaneously with desorption of DIMP from the reactor's walls, which partially compensates for the loss of DIMP from the gas-phase.
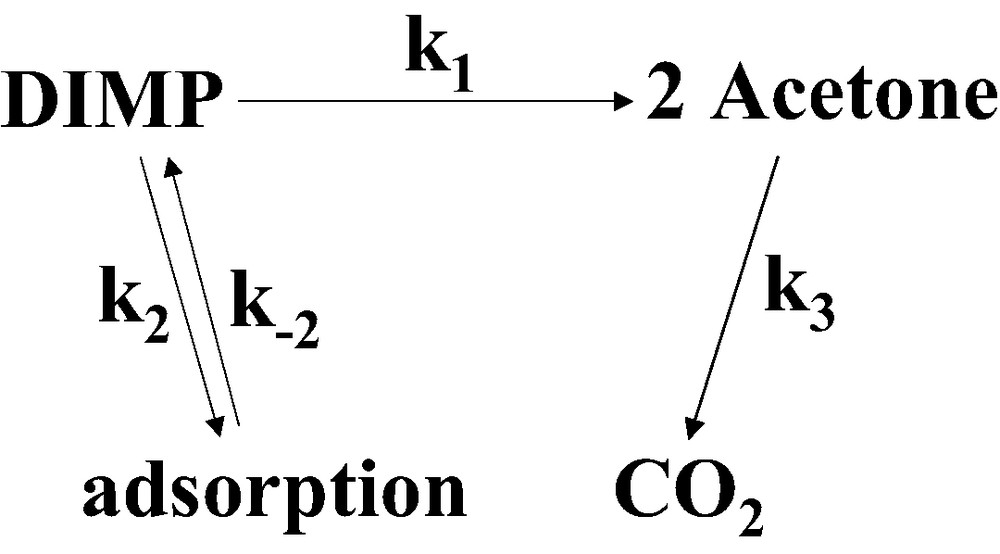
The photodegradation scheme for DIMP.
The effect of the MUACu MRS on the photodegradation of DIMP is demonstrated in Fig. 6, which shows the kinetics of formation of acetone for both types of substrates, based on the area of the acetone-related 1735 cm–1 peak. As depicted in the figure, the rate of production of acetone in the presence of MUACu domains is higher by a factor of 2.5 than that observed with samples containing the same photocatalytic surface area but without MUACu. Similarly, examination of the production of CO2 in these two systems revealed a faster increase in the mineralization rate for the MUACu-containing system, albeit by a factor of less than three. It is known that Cu2+ and some of its chelates may act as hydrolyzing catalysts for phosphonates [62]. It is noteworthy therefore, that exposure of DIMP to these two systems in the dark did not yield any significant amounts of acetone or CO2.
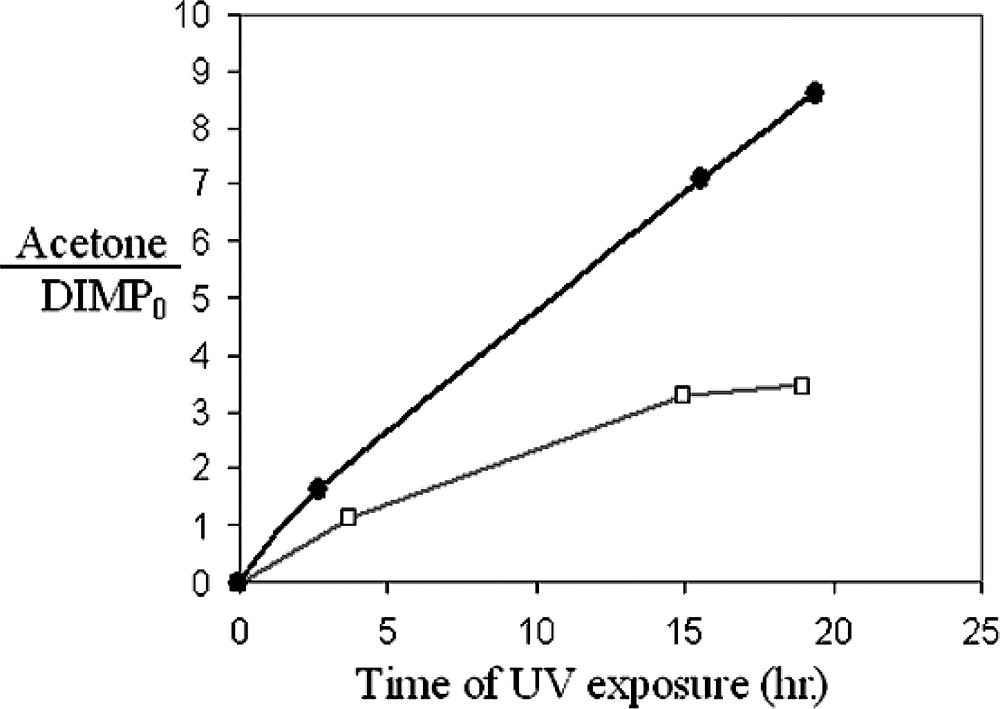
The kinetics of acetone production during photodegradation of DIMP on structures made of 5 μm width stripes of TiO2/Au: without MRS, •: with MRS.
In order to find out whether the described-above enhancement in the quantities of the intermediate product acetone in the MRS-containing system was due to enhanced degradation of DIMP by the MUACu or due to slower photodegradation of acetone, the photodegradation of acetone was measured with various substrates. These substrates included structures made of stripes of TiO2 (5 μm in width) and gold (5 μm in width), structures made of stripes of TiO2 (5 μm in width) and gold (5 μm in width) coated with MUACu and thin films of titanium dioxide without any metal stripes. Care was taken to have the same active area in all three systems. Plotting the integrated absorbance of the acetone peaks versus time yielded an exponentially decaying curve (R2 > 0.99), suggesting apparent first order kinetics. For comparison, a first order reaction rate constant, deduced from the acetone degradation kinetics measured during the photodegradation of DIMP, but at very long exposure times (i.e. once all DIMP was consumed), was also measured. The rate constants of these four systems were found to be identical (0.013 ± 0.001 h–1), demonstrating that the presence of MUACu or gold do not have any effect (neither positive nor negative) on the rate of acetone photodegradation. Moreover, the kinetic rate constant of the photodegradation of acetone that was produced from DIMP was similar (0.014 h–1). Hence, it can be concluded that this structure enhances specifically the degradation of DIMP but not that of acetone.
In order to quantify the benevolent effect of the ‘adsorb-and-shuttle’ approach, it is essential to calculate the extent by which the photodegradation rate of DIMP is improved by this approach. Taken that the contaminant is adsorbed reversibly on the reactor's walls, and assuming quasi steady state with respect to DIMP adsorbed on the reactor vessel, it is possible to calculate the ratio between k1 (see Fig. 5) in a system containing the MRS and k1 in the control system. This ratio can be calculated based on the IR kinetics of any of the DIMP peaks, or based on the rise and fall of the acetone peaks (Eq. (1)). The latter way takes into consideration the adsorption/desorption of DIMP from the reactor walls. Values of 0.16 and 0.04 h–1 were calculated, based on fitting the kinetics of the degradation of DIMP, whereas values of 0.1 and 0.01 h–1 were calculated based on the acetone kinetics (Fig. 7). This reflects an improvement in k1 by a factor of 4–10 due to the presence of the molecular recognition sites.
| (1) |
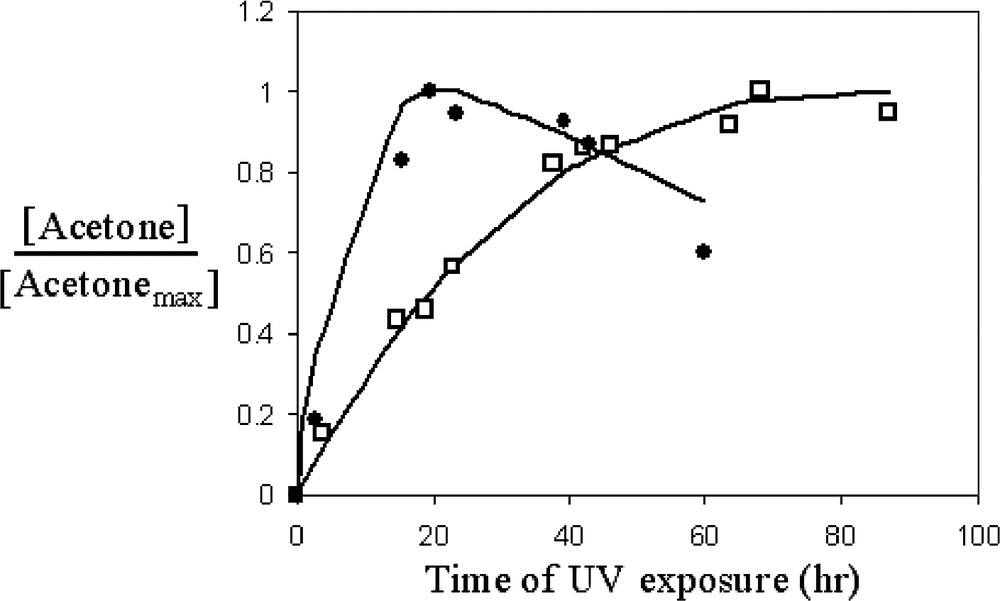
The kinetics of acetone production as fitted according to Eq. (1). □: without MRS, •: with MRS.
Unlike the thiolated β-cyclodextrin system, some degradation of the organic part of the MRS upon irradiation was observed. This could put an apparent limitation on the use of these structures in practical devices. Nevertheless, repeated measurements done following a 2-h recovery time in air did not find any long-term reduction in the photoefficiency of DIMP degradation, in comparison with the first run. XPS measurements suggest that in that case, the thiol part of the MRS remains attached to the metal and is connected to the copper ion, which is the binding site for the DIMP.
In conclusion, an improvement by a factor of app. 4–10 was obtained for a structure, made of alternating stripes of titanium dioxide and molecular recognition sites on gold, both having 5 μm in width. It is sensible to believe that much greater improvement is possible in powders, where the average diffusion length is much smaller than the 1.3 μm in the present experiments. Since the affinity between MUACu and DIMP relies on the phosphonate group, which is common to DIMP and Sarin, it is expected that this system will be suitable also for Sarin.
The fact that the benevolent effect of the MUACu was noticed with DIMP, but not with acetone, evidently shows that MRS may serve not only to increase degradation rates but also to induce preferential photodegradation. Such selectivity is of great importance in particular in systems where the intermediate products are far less hazardous than the initial reactants, as indeed happens in this system, and may also happen in the photodegradation of many chemical warfare agents. The results presented in this work point also to the need for better protection of the molecular recognition sites from photodegradation, and for better understanding of the effect of copper ions on the reaction rate.
3.6 Meso-porous materials
Selection by size can be achieved by developing photocatalytic materials that contain well-defined specific pores. As an example, one may outline the incorporation of titanium dioxide into sheet silicates of clay [63]. Here, the distance between adjacent layers in the clay may act to enhance the photodegradation of molecules that are smaller than the interlayer distance. Indeed, when a comparison of the rates of photodegradation of a series of carboxylic acids on TiO2–clay structures and on TiO2 powder was done, it was found that small molecules (up to C8) degraded faster by the composite structure, whereas capric acid (C10) degraded faster by the TiO2 powder. It should be mentioned, though, that other parameters could also contribute to the enhancement in the photodegradation of small molecules by the TiO2–clay composite, for example, a shift in the location of the bottom of the conduction band of the embedded titanium dioxide particles. The difference between the relative adsorption on TiO2 and on TiO2–pillared clays (mica, montmorillonite and saponite) manifested itself also in the distribution of species that were produced during the photodegradation of benzene [64].
Treating of a microporous titanosilicate photocatalyst with HF was found to increase both its activity and its selectivity. This way, in a mixture containing phenol and 2,3-dihydroxynaphthalene (DHN), a complete abatement of the DHN was observed within 10 min, while phenol concentration was basically unmodified [65]. It was suggested that the zeolitic internal cavities offer a protective environment against degradation for species that can easily diffuse inside. Alternatively, this size selectivity could originate from the ability to accommodate contaminants in the vicinity of the pore mouth, at the end of the Ti–O–Ti chains.
Another evidence for altering the specificity was demonstrated when nanoparticles of titanium dioxide were grafted onto the pore surface of meso-porous silicates. The degradation rate of α-terpinol was enhanced by a factor of 4 compared with P25, while that of the dye Rhodamine-6 G was reduced by a factor of 2 [66].
3.7 Imprinting
Another way to obtain high reactant selectivity is to imprint [67] cavities of the target molecules on the photocatalyst's surface. Within the context of preferential photocatalysis, imprinting means the sol–gel polymerization of titanium dioxide around a molecule acting as a template (Fig. 8). After polymerization, the molecular template is chemically or physically removed, to leave the imprinted molecular site. Here, a promising method was proposed by Lee et al. [68] according to which a Ti(IV) alkoxide was modified by reacting with a carboxylic acid derivative of the target molecule. This method was successfully adapted recently to give high selectivity in the photodegradation of 2,4-dichlorophenoxyacetic acid [69]. Recently, we have imprinted DIMP onto titanium dioxide. Although imprinting of these molecules did not induce so far higher degradation rates in comparison with non-imprinted TiO2, a complete mineralization was observed, without the appearance of any intermediate acetone, thus suggesting that the molecular cavities serve to trap the contaminant until full mineralization takes place. Overall, the imprinting approach, although in its infancy, seems to be very promising due to its simplicity, for those contaminants that can bound directly to TiO2 precursors, either without or with a linker.
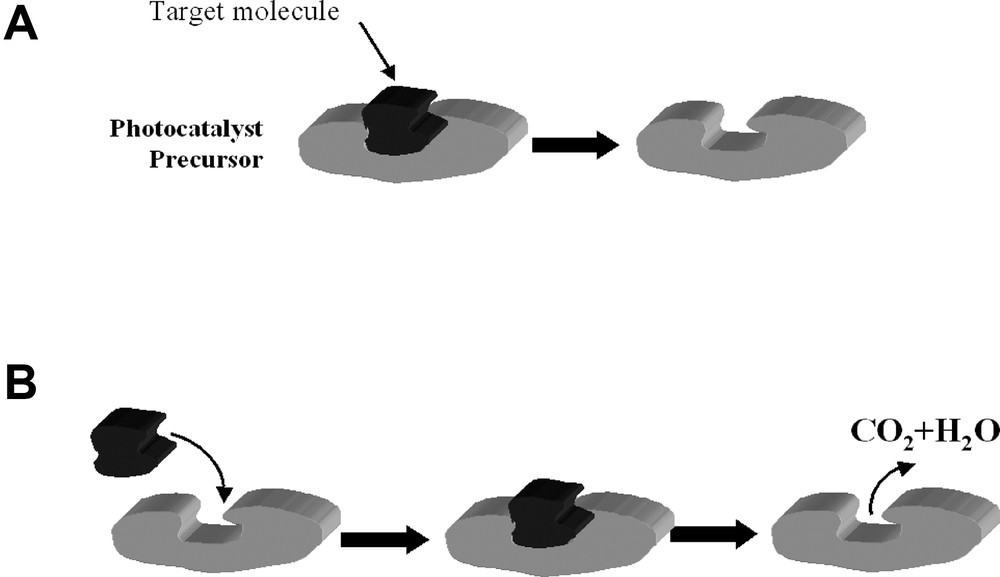
The imprinting scheme. (A) Preparation of the photocatalyst. (B) Using the modified photocatalyst.
4 Mixed waste vs. non-mixed waste streams
In Sections 2 and 3, several approaches that may bring to preferential photodegradation were presented. In the absence of documentation on the effects of these approaches in multicomponent streams, their analysis was based upon comparing the measured performance under a single component situation. Although such a comparison is somehow limited, it is still valuable, provided that the differences between comparing several single component streams and obtaining information from multicomponent streams are taken into consideration. These differences are discussed herein.
The photodegradation rates in mixed streams often reflect a competitive adsorption situation. Hence, relative rates that are measured based on single component experiments of various contaminants should be regarded as qualitative guidelines for each approach and not as predictive values for mixed stream situations, as indeed was found for a mixture containing oxalic acid, formic acid and formaldehyde [70]. As a rule of thumb, the degradation rates of strongly adsorbed contaminants are not expected to vary by much in the presence of other contaminants, whereas, the degradation rates of contaminants that adsorb weakly are expected to change significantly in the presence of other contaminants.
The difference between mono and multi component streams can also be the consequence of the formation of intermediates, or end products, that influence the degradation of cross contaminants. As an example, one may outline the cross effect of chlorides, produced during the photodegradation of 2-chlorophenol, on the degradation of 2-nitrophenol, and the effect of nitrate ions produced during the degradation of 2-nitrophenol on the degradation of 2-chlorophenol [20].
It is known that the degradation of contaminants that are not protonated (such as 2-chlorobiphenyl) is hardly affected by pH, whereas the degradation of contaminants that can be protonated (such as formic acid or acetic acid) is affected. Nevertheless, pH changes in mixed waste streams were found to affect the degradation rates of both types of molecules [16]. This effect, manifested by an increase in the pseudo first-order rate constant for the degradation of 2-chlorobiphenyl, from 0.4 min−1 at pH 3.8 to 2.6 min−1 at pH 6, was attributed to the varying role of competitive adsorption in the mixed- stream case.
The photocurrents that are produced during the illumination of biased TiO2 in an aqueous solution containing mixed contaminants (methanol + oxalate, methanol + salicylate, oxalate + salicylate) were measured by Calvo et al. [71]. A non-symmetric effect was claimed. Hole scavenging and electron injection by a weakly adsorbed species (such as methanol) was suppressed by the presence of chemisorbed photolytes (such as salicylate and oxalate), whereas the degradation pathway of chemisorbed photolytes was hardly influenced by the presence of weakly adsorbed photolytes.
A thorough study of photodegradation of as many as 14 binary mixtures was presented by Lichtin et al. [72]. Various inhibition effects were observed among which were a strong inhibitive effect of CH3OH on CH2Cl2, a mild inhibitive effect of CH2Cl2 on CH3OH, a promoting effect of CCl4 on the removal of methanol, an insignificant effect of methanol on the rate of CCl4 removal, a mutual inhibitive effect of CH3OH and trichloroethylene, and an inhibitive effect of TCE on the removal of acetonitrile. In a different work, the same group found that addition of 0.03% of TCE to 0.1% i-octane-containing air increases the photodegradation rate of the octane [73]. A similar effect, this time on the photodegradation of toluene, m-xylene, ethybenzene (but not benzene) was found by Luo and Ollis [74] and d'Hennezel and Ollis [75]. Here, the synergetic effect was explained by the presence of chlorine radicals, formed in the photocatalytic degradation of gas-phase TCE. These radicals are also considered to be the source for the very high quantum yields that were reported for the photodegradation of gas-phase TCE [4]. It is noteworthy that the enhancive effect of TCE on the degradation of toluene was not observed on composite particles composed of P25 and activated carbon, although it was confirmed in non-composite titanium dioxide [76].
5 Product selectivity
Although this manuscript is aimed at analyzing preferential photodegradation, it is beneficial to examine a few cases of product selectivity, since the two issues are interconnected, through the fact that the intermediates often compete with the reactants on the precious active species (OH radicals, holes, electrons, etc.). Unlike the degradation of contaminants in mixed streams, where data on preferential degradation is quite scarce, there are quite a few manuscripts on the effect of various parameters on the type of products, probably because of the direct economical benefit that was expected upon photocatalytic synthesizing of desired molecules. Although a detailed summary is beyond the scope of this manuscript, it can be claimed that, basically, the same parameters that affect preferential photocatalytic degradation seem to influence product distribution. This is due to the fact that in most cases, product selectivity was governed by the adsorption of the intermediates. Such parameters include: pH (for example: the degradation of methanol [30]), zeolite structure (for example: the degradation of NO to N2 [77]), surface derivatization (monooxygenation of cyclohexane [78]), and the kind of solvents that are used [79,80].
6 Conclusion
Despite the relatively scarce data on preferential photodegradation, it is possible to outline several process parameters, such as pH, doping, and type of solvent that may provide some specificity. Most of these parameters act through controlling the mass transport of the reactants to the photoactive surfaces. Mass transport control is also the basis for several synthetic approaches to obtain specificity, out of which surface modification by imprinted contaminants, and the ‘adsorb-and-shuttle’ approach should be considered as methods that may provide high specificity. Due to the cross-influence between co-existing contaminants, it is recommended to concentrate on multicomponent streams when studying this issue.