

1 Introduction
The field of organic electronic devices, such as light-emitting diodes [1], field-effect transistors [2], sensors [3], lasers [4] and photovoltaic devices [5] is rapidly expanding due to the promise of inexpensive material and device fabrication accompanied by the wide variety of functionality of organic materials. The most important functionality is the large polarizability of the extended π-conjugated electron systems of organic compounds, among which π-conjugated semiconducting polymers are one of the prominent materials [6]. The semiconducting properties of conjugated polymers are attributed to the dimerization (i.e. Peierls distortion) of double bonds formed by the delocalization of the pz-orbitals of the carbon atoms of the polymer backbone [7].
Another interesting class of functional materials is fullerenes. After two decades from the initial discovery of buckminsterfullerene by Kroto et al. [8] in 1985, these materials are being developed to various applications as antioxidants, biopharmaceuticals and active and passive additives for the polymer industry. The science and technology of plastic solar cells which will be reviewed here lies on the frontier of the above mentioned two important classes of functional materials, and shows an example how interdisciplinary research fields can merge together towards the developments of future technologies.
This review paper is organized as follows: After a brief history of organic solar cells, the photophysical properties, the spectroscopic observation of ultrafast photoinduced electron transfer between conjugated polymers and fullerene, will be reviewed. Then the device concept of bulk heterojunction will be introduced together with an overview of the current research of plastic solar cells, focusing primarily on novel materials and advanced characterization techniques. Finally as an outlook for future developments, the emerging technologies of inorganic-organic hybrid photovoltaic devices will be introduced.
2 En route to bulk heterojunction solar cells
Although the idea of using organic materials for converting solar energy into electrical power is dated back to the development of organic photoconductor materials for xerography and printing technology, it was in 1985 when Tang [9] has demonstrated an organic solar cell with power conversion efficiency around 1%. This cell was based on a doublelayer-structure of two well known photoconductors Me-Ptcdi (N,N′-dimethyl-perylene-3,4,9,10-dicarboximide)/ZnPc (zinc–phthalocyanine), both of which are rather inefficient photovoltaic materials when used in single layer structure. This is because charge generation efficiency in pure organic materials is very low attributed to the small mean free path (low mobility) of charge carriers, which is smaller than the Coulomb radius (small dielectric constant) [10]. Therefore primary photoexcitations are strongly bound neutral excited states (binding energy in excess of 0.5 eV), and the generation of free charge carriers requires excess energy gained by for e.g. electron transfer from the donor containing the excitation to lower lying molecular orbital of the electron acceptor moieties or to interfaces. The device concept of multilayer small molecule photovoltaic devices has been pursued until today, and power conversion efficiency around 3.5% in laboratory scale has been presented [5]. In the meantime, conductive polymers gained interest due to the above mentioned semiconducting properties of conjugated π-electron systems accompanied by easy processing and flexibility of common plastics. More specifically, the experimental observation of photoinduced charge transfer between the photoexcited state of a non-degenerate conjugated polymer to buckminsterfullerene [11] by various spectroscopic techniques stimulated intensive research, which is briefly summarized below:
- • mixing only a few mol percent of PCBM with a strongly luminescent conjugated polymer MEH-PPV, the photoluminescence is efficiently quenched, and the steady-state photoconductivity is increased (Fig. 1) [12];
- • steady-state photoconductivity experiments (PC) showed that blending conjugated polymers with only 1% C60, the PC increased by orders of magnitude. From time resolved photoconductivity experiments it was concluded that the increased steady state photoconductance is the cumulative effect of increased charge generation and increased charge carrier lifetime. (Fig. 2) [13];
- • instead of the single band centered around 1.35 eV in the photoinduced absorption (PIA) of pristine MEH-PPV, a sharp PIA edge at 1.1 eV and a plateau at 1.6 eV–2.0 eV is observed in the MEH-PPV:C60 blend [14] (Fig. 3). From photoinduced-absorption-detected magnetic resonance [15], the 1.35 eV peak in the pristine polymer has been attributed to triplet–triplet absorption, which is completely quenched in the blends with C60. This observation is consistent with a photoinduced charge transfer, which is sufficiently fast to quench singlet to triplet intersystem crossing in MEH-PPV resulting in absorption features attributed to positive radical cations (photodoping) [12]. Subpicosecond transient absorption studies have revealed that the new absorption bands appear within 300 fs upon photoexcitation, and remain almost unchanged for longer time delays [16]. More recent transient absorption studies showed that the photoinduced charge transfer happens within 40 fs after photoexcitation, quenching the coherent vibrational excitations of the conjugated polymer, therefore its quantum yield approaches unity being faster than any other excited state relaxation mechanisms [17];
- • definitive evidence for the occurrence of photoinduced charge transfer was obtained by light induced ESR (LESR) studies [11,18]. Upon photoexcitation of the MEH-PPV: C60 mixture with energy larger than the π–π* band of the conjugated polymer, two ESR signals are detected. The one with a g factor value < 2 is characteristic of a C60•– radical anion, meanwhile the g ~ 2.002 value is typical of a conjugated polymer radical cation. Time-resolved LESR studies also showed that the signature of the charges may remain detectable for hours indicating persistent photoconductivity at low temperatures [19];
- • transient absorption studies in the ns–ms time regime showed that the photogenerated charges created are long living with a power law decay in the long time scale indicating that some a portion of the charge carriers may be deeply trapped [20].
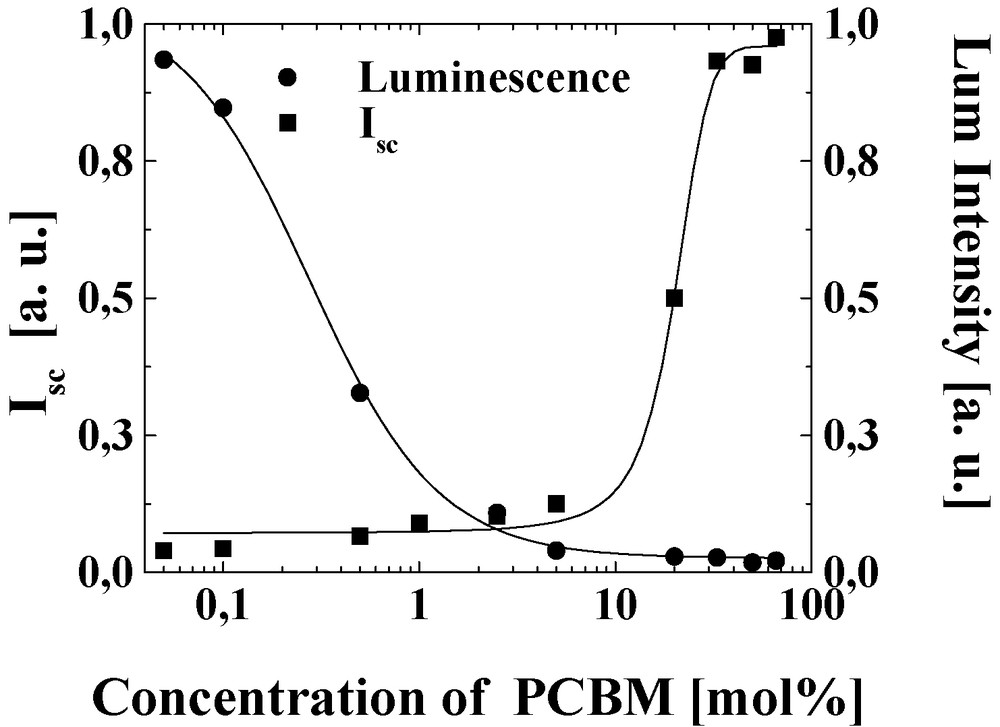
Photoluminescence quenching (•) and short circuit photocurrent (■) of a blend of MEH-PPV: PCBM as a function of the concentration of PCBM. The Pl of MDMO-PPV is efficiently quenched by adding a few mol% PCBM; however the photocurrent increase occurs at higher PCBM load corresponding to a percolation threshold.
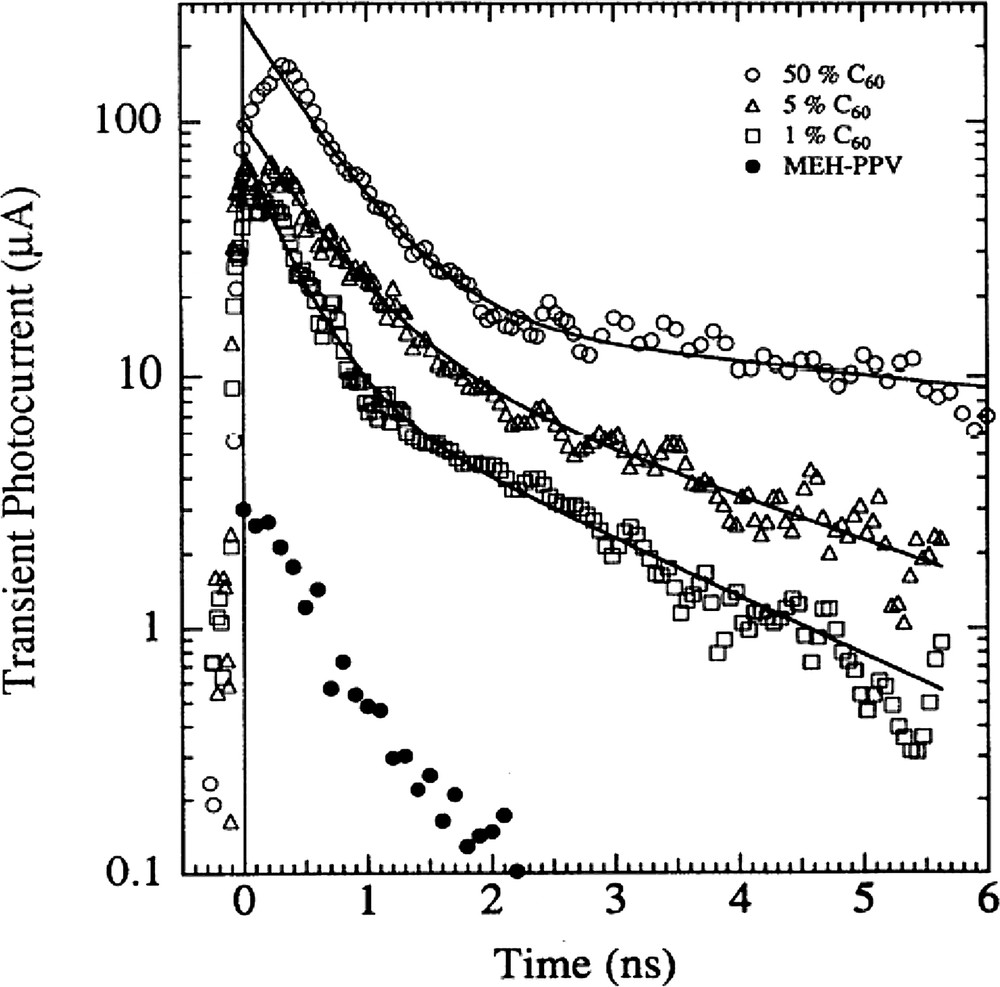
Transient photoconductivity of (•) pristine MEH-PPV, and its various blends with C60. Upon blending MEH-PPV with C60, the peak photoconductivity is increased by orders of magnitude accompanied by an increase of the lifetime of the photogenerated charge carriers. Reprinted with permission from [13]. Copyright (1993) by the American Physical Society.
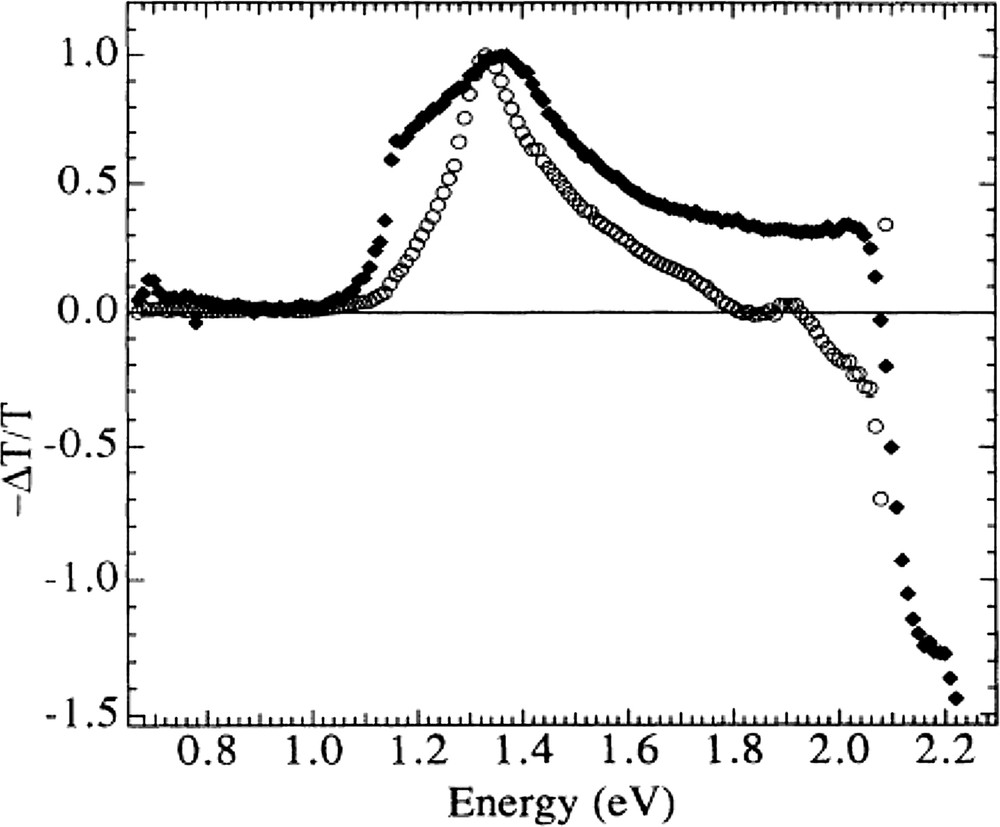
Photoinduced absorption (PIA) spectra of MEH-PPV (○) and MEH-PPV: C60 composite (•) at 80 K obtained by pumping with an argon ion laser at 2.41 eV with 50 mW. Reprinted with permission from [14]. Copyright (1993) by the American Physical Society.
The above experimental observations are consistent with an ultrafast, photoinduced electron transfer between the photoexcited state of the conjugated polymer and fullerene creating metastable charges with micro to milliseconds lifetime. An immediate realization was that the application of selective electrical contacts to the above described system, an efficient photovoltaic device may be fabricated. First, bilayer devices have been presented exhibiting short circuit photocurrent of a few μA cm–2, and monochromatic power conversion efficiency around 0.1% [21]. The real breakthrough was achieved by creating interpenetrating network of the electron donor and electron acceptor materials resulting in a device architecture often referred to as the bulk heterojunction [22].
3 Bulk heterojunction solar cells
3.1 Concepts of device operation and fabrication
The concept of a bulk heterojunction is shown in Fig. 4 together with a band structure of a typical device displaying the relevant energetic levels (highest occupied molecular orbital (HOMO) of the electron donor, lowest unoccupied molecular orbital (LUMO) of the electron acceptor. The chemical structure of some materials used in organic solar cells is exhibited in Fig. 5.
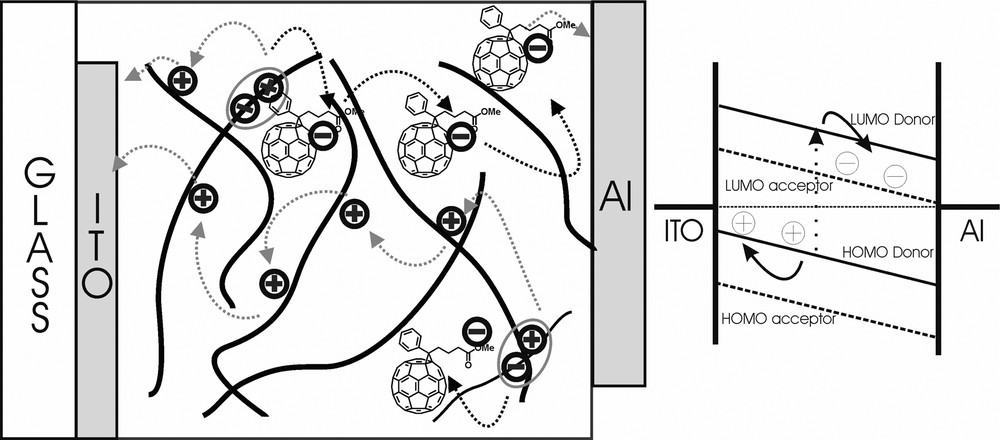
Schematic illustration of a bulk heterojunction based on the interpenetrating network of a conjugated polymer and PCBM. Charge carriers generated by photoinduced electron transfer are moving towards the corresponding electrode. The electronic levels relevant to the device operation are also displayed.

Chemical structures of materials used in the Tang solar cell: Zink-phthalocyanine and Me-Ptcdi (perylene-3,4,9,10, tetracarboxylic-N,N′-dimethyle-diimide), and in plastic solar cells: MDMO-PPV poly[2-methoxy-5-(3,7-dimethyloctyloxy)-phenylene vinylene], P3HT (poly(3-hexylthiophene-2,5-diyl), PCBM 1-(3-methoxycarbonyl)propyl-1-phenyl-(6,6)-C61, and PTPTB (poly-N-dodecyl-2,5,-bis(2′-thienyl)pyrrole,2,1,3-benzothiadiazole.
The charge carriers are generated by photoinduced electron transfer from the LUMO of the electron donor (conjugated polymer) to the LUMO of the electron acceptor (PCBM). For efficient charge generation, an exciton photogenerated anywhere in the blend has to reach an acceptor site within its lifetime; therefore the magnitude of the maximum allowed phase separation is determined by the parameter called exciton diffusion length. For intimately mixed blends, experiment showed that only a few weight% of the electron acceptor is sufficient to quench virtually all excitations (Fig. 1). For efficient photovoltaic devices, however, the created charge carriers need to be transported to the corresponding electrodes within their lifetime, which will be a strong function of charge carrier mobility of the materials. The ability of transporting both charges in a blend of donor and acceptor materials will be also a strong function of morphology, i.e. creating interpenetrating, bi-continuous network of both materials. For e.g. nearly 20% load of PCBM, corresponding roughly to a percolation threshold for a 1D mixed in a 3D system, is necessary for obtaining significant photocurrent increase (Fig. 1).
As a last step, charge carriers are extracted from the device through two selective contacts. A transparent indium tin oxide (ITO) coated glass matching the HOMO level of the conjugated polymer (hole contact) is used on the illumination side, and evaporated thin (80–100 nm) lithium fluoride/aluminum metal contact matching the LUMO of PCBM (electron contact) is used on the other side. In the state-of-the-art devices, a thin (100 nm) hole-injection layer, a highly doped PEDOT-PPS film is used, which also serves to smoothen the surface of the ITO and to increase device stability. The maximum open circuit voltage of an MDMO-PPV: PCBM device is ~0.8 V at room temperature, which corresponds to the energy level difference between the LUMO of PCBM, and the HOMO of MDMO-PPV. Moreover, it was observed that the open circuit voltage is only a very weak function of the work function of the cathode material used, but strongly depends on the LUMO of the electron acceptor used [23].
The organic layers of such a cell are deposited by solution processing techniques, such as spin coating [24], doctor blading [25], screen printing [26] and more recently, by spray coating [27]. The ultimate goal from a technological point of view is a roll to roll printing production of flexible plastic solar cells in which process no vacuum evaporation step is necessary.
From the above description it is realized that the main issues of improving solar cell efficiency are (i) the optimization of the nano-morphology, i.e. to understand and control parameters influencing the phase separation in the photoactive blend, (ii) the determination and improvement of the charge carrier mobility and (iii) improving the spectral sensitivity, for e.g. by using low bandgap conjugated polymers. These research aims will be reviewed next.
3.2 Morphology of bulk heterojuncion solar cells
The power conversion efficiency of bulk heterojunction solar cells based on MDMO-PPV: PCBM 1:4 mixture could be improved from approximately 1% (AM 1.5) to 2.5% by simply changing the solvent from which the active layer has been cast [24]. Combined atomic force microscopy (AFM) and scanning electron microscopy (SEM) investigations [28] showed that large PCBM clusters are formed in the toluene cast films, meanwhile a smoother morphology corresponding to well intermixed system is observed in the chlorobenzene cast films [29]. Fig. 6 shows cross-section SEM pictures of the films cast from different solvents [30]. In the case shown on the left, spheres with diameters around 20 nm are embedded in a matrix, meanwhile in the toluene case (right), large clusters are formed. A recent study combining AFM, transmission, cross section SEM and thermal annealing experiments has clearly correlated the rather complex morphology of bulk heterojunction solar cells to the device performance [29,30].

Cross section SEM images of MDMO-PPV/PCBM 1:4 weight ratio blend spin coated from toluene (right) or chlorobenzene (left) solutions.
3.2.1 Donor–acceptor ‘double-cable’ polymers
The above mentioned examples showed that although the preparation of bulk heterojuncton solar cells is very simple, the proper control of the processing parameters is rather complex, and involves detailed studies of for e.g. dependence of the device performance on the choice of solvent, the concentration of the solution, and the weight ratio of the active components. In order to have a control on the morphology at the molecular level, the concept of ‘double-cable’ polymers has been introduced [31] (Fig. 7). Ideally, phase separation in these materials is prevented by attaching the electron acceptor moieties directly to the conjugated backbone by covalent bonds. It is expected that the electron created by photoinduced electron transfer will travel by hopping between the acceptor moieties; meanwhile the rather high on-chain mobility of the remaining hole on the conjugated chain can be utilized in transporting the positive charge. Several of these double-cable polymers has been developed exhibiting interesting photophysical properties, yet the real breakthrough from the efficiency of the photovoltaic device point of view has not been achieved probably due to inefficient interchain transport [32].

Schematic representation of ‘double-cable’ polymers. The charge carriers generated by photoinduced charge transfer can be in principle transported within one molecule, therefore termed as a ‘molecular heterojunction’.
3.3 Charge-carrier mobility in bulk heterojunction solar cells
The drift distance of the charge carriers generated anywhere within the photoactive layer can be calculated as , where τ is charge carrier lifetime, E is the electric field and μ is the mobility. This equation is based on the assumption that the charge carriers are electric field driven [33]. Charge carrier mobility μ is an important parameter limiting the maximum thickness of the active layer as lmax < ld. Charge-carrier mobility in bulk heterojunction solar cells has been studied by a time of flight (ToF) technique [34], or calculated from the transfer characteristic of a field-effect transistor (FET) [35]. These results indicated that the electron (PCBM phase) and the hole (conjugated polymer phase) mobility in the photoactive blend is fairly balanced, which is counterintuitive to experiments performed on the pristine materials, which showed that the electron mobility of thin films of PCBM is approximately 4000 times higher that the hole mobility of pristine MDMO-PPV films [36].
The latter results indicate, that the hole mobility of the pristine MDMO-PPV needs to be improved. One viable way to improve charge carrier mobility in conjugated polymers is to increase the regioregularity of the conjugated backbone. Lutsen et al. has reported a regio-controlled synthesis of MDMO-PPV via the sulfinyl precursor route [37]. Unfortunately, the fully regioregular MDMO-PPV is insoluble, which prevents otherwise promising substances from the application in bulk heterojunction solar cells. However, a soluble regiospecific MDMO-PPV has been synthesized via the sulfinil precoursor based on the 70:30 weight percent mixture of Monomer A and Monomer B, as illustrated in Scheme 1 [38].
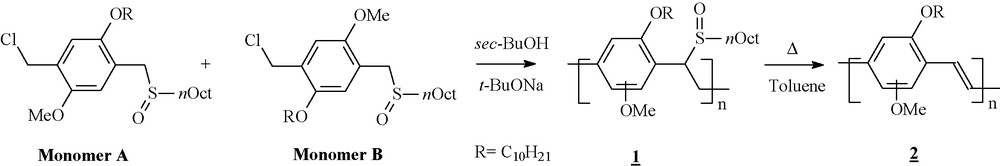
Synthesis of the MDMO-PPV copolymers.
The room temperature ToF mobility of this novel polymer (70:30 RS-MDMO-PPV) is found to be ~3.5 times higher at all electric fields as compared to the commercially available regiorandom RRa-MDMO-PPV (Fig. 8). From temperature and electric field dependence of mobility studies, the improved mobility at room temperature has been attributed to the presence of ordered regions embedded in an otherwise amorphous polymer matrix [38]. The improved charge-transport properties of the novel regiospecific MDMO-PPV polymer has been utilized in fabrication of photovoltaic devices with enhanced power-conversion efficiency, mainly due to the high (0.7) filling factor (Fig. 9).
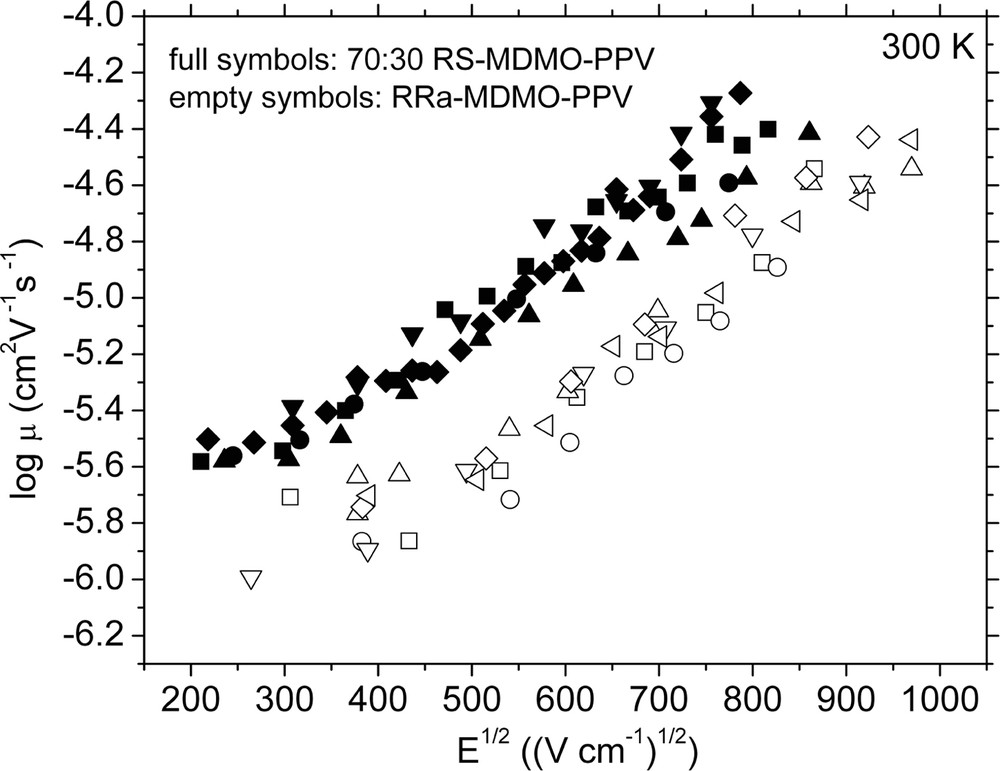
Charge carrier mobility of 70:30 RS-MDMO-PPV (full symbols) and RRa-MDMO-PPV (empty symbols) determined by a time of flight technique at room temperature.
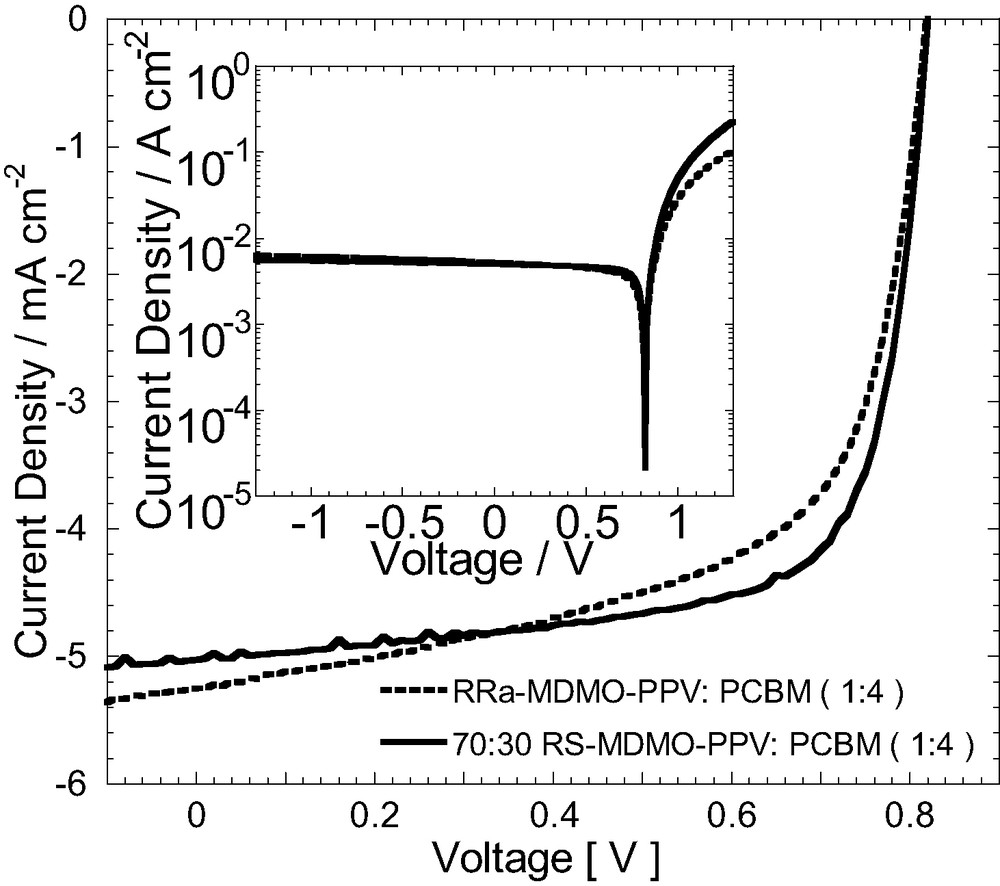
Current–voltage curves of MDMO-PPV: PCBM bulk heterojunction solar cells under simulated AM 1.5 illumination. The filling factor defined as FF = (Impp × Vmpp)/(Isc × Voc) is 0.71 for the 70:30 RS-MDMO-PPV, which is attributed to the increased charge-carrier mobility.
A more drastic improvement of the performance of bulk heterojunction solar cells [39] has been achieved by using regioregular poly(3-hexylthiophene) (P3HT), a well soluble conjugated polymer exhibiting the highest charge carrier mobility reported up to date among conjugated polymers [1].
We have investigated the charge transport properties of P3HT by the ToF technique, and observed a negative electric field dependence of mobility first time in conjugated, semiconducting polymers [40]. Fig. 10 shows the measured mobility versus squared electric field at various temperatures. The logarithm mobility follows the typical Poole–Frenkel-like log μ∝βE1/2 field dependence, β > 0 at lower temperatures, yet with increasing temperatures the field dependence diminishes, and eventually turns to negative above ~250 K. Such negative electric field dependence of mobility has been observed in several disordered charge transport materials, such as molecularly doped polymers, and molecular glasses, and has been analyzed in the framework of disorder formalism [41] (Eq. (1)) developed by Bässler and coworkers.
| (1) |
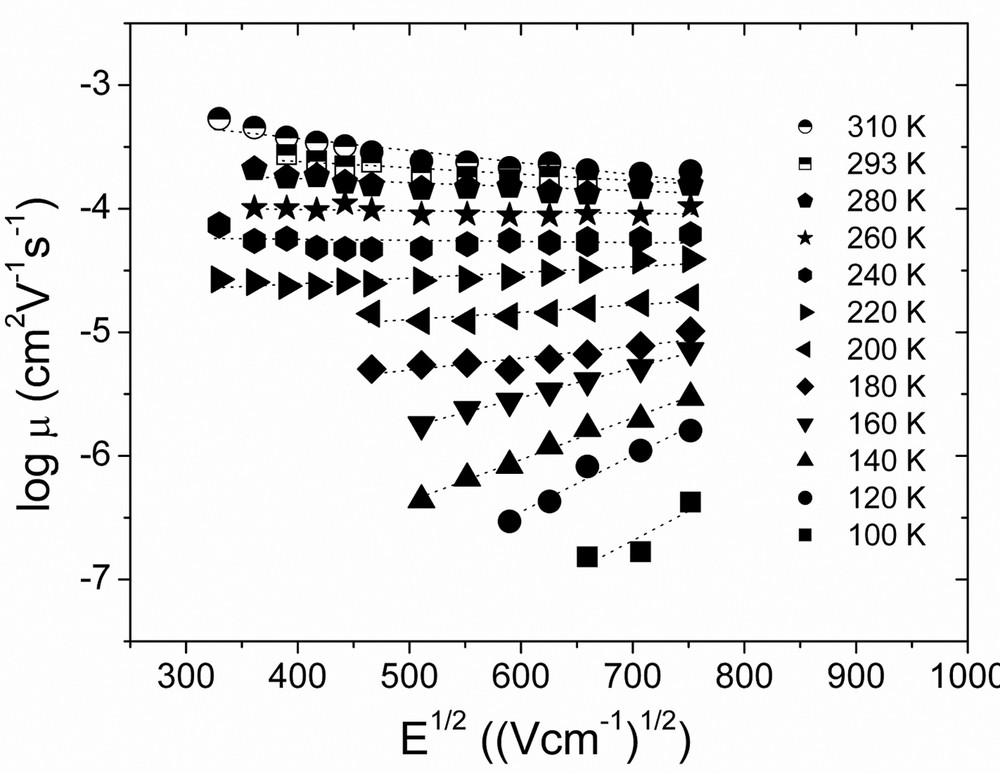
Electric-field dependence of the hole mobility in regioregular P3HT at various temperatures determined by the time-of-flight technique. The lines represent linear fits of the data.
3.4 Enhanced photonharvesting
The spectral photon flux under AM 1.5 test conditions is compared to the absorption spectra of the MDMO-PPV: PCBM blend in Fig. 11. An immediate realization is that the absorption of the photoactive blend is very limited in spectral regions where the spectral photon flux from the sun is the most intense. The onset of the absorption of the conjugated polymer corresponds to the π–π* absorption, and in order to extend the spectral sensitivity of bulk heterojunction solar cells, materials with lower π–π* band gaps are required. There have been significant research efforts in synthesizing such low bandgap polymers [43] and the results for one successful candidate PTPTB [44] (see structure in Fig. 4) is summarized. The absorption of this polymer is shown in Fig. 11 for comparison. The onset of the absorption is found around 780 nm, corresponding to an optical bandgap of ~1.6 eV, at which the terrestrial solar spectra is more intense. Mixing this polymer with PCBM, photovoltaic devices with power conversion efficiency of 1% has been achieved. Spectrally resolved photocurrent measurements at short circuit conditions (incident photon to converted electron, IPCE) showed that the IPCE follows closely the absorption of the conjugated polymer. The rather low filling factor is mainly responsible for the rather limited performance of these devices, which indicates that changing one parameter (bandgap) may influence other properties, such as charge carrier mobility, morphology of the blend, or the contact formation properties. To optimize all these parameters comprise a rather complex and challenging task for material scientists.
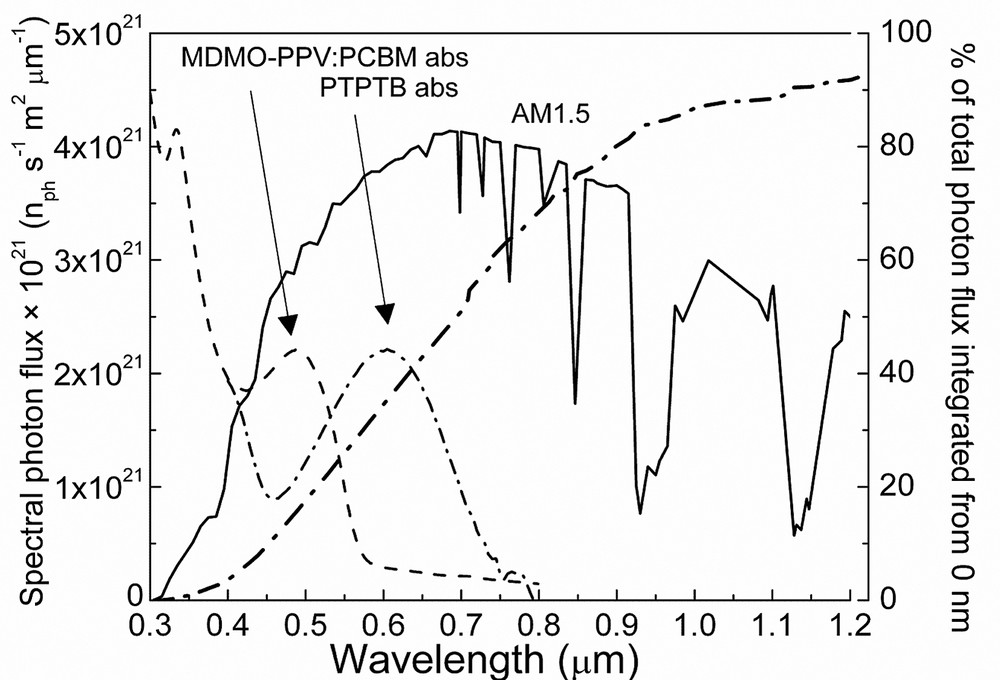
Spectral photon flux and integrated total photon flux of AM 1.5 solar spectra. The absorption of MDMO-PPV: PCBM blend and the low bandgap polymer PTPTB is displayed for comparison.
4 Alternative routes. Inorganic–organic hybrid solar cells
The state of the art MDMO-PPV: PCBM bulk heterojunction solar cell exhibit external quantum efficiency nearly 60% at the absorption maximum, and comparison with the fraction of absorbed light at this wavelength shows that the losses limiting device efficiency are mainly optical [45]. The main reason is that fullerenes have only very weak absorption in the visible spectral range; therefore the main component of the photoactive blend is actually almost optically inactive. To overcome this limitation, alternative electron acceptors that are capable of absorbing visible light preferably at the lower energy part of the spectra are required. Moreover, the strong optical absorption needs to be accompanied by efficient electron transport. Various inorganic n-type semiconductors, such as CuInS2, CuInSe2, CdSe, CdTe, GaAs etc. are known photovoltaic materials when used as bulk or thin films. It was recently shown that nano-sized particles of these semiconductors can be synthesized by wet chemical synthesis. The obtained nano-sized particles exhibit unique properties different from that of the bulk semiconductor, for e.g. tunable color which depends on the size of the nano-crystals (quantization effect). When these inorganic nanoparticles are mixed in a semiconducting conjugated polymer host, a photovoltaic device (hybrid solar cell) can be fabricated with external quantum efficiency of 60% [46]. The main components of such hybrid solar cells are generally incompatible, therefore significant effort is focused on improving the miscibility of the components [47]. An advantage of using the hybrid solar cell approach is the well known properties of inorganic semiconductors, the possibility of tailoring the properties by inexpensive wet chemical synthesis through the quantization effect and the almost unlimited choice of inorganic/organic material combinations. As a demonstration, one promising system based on CuInS2 embedded in various organic matrixes [48] has been recently reported.
The above examples of using organic materials demonstrated that the bulk heterojunction concept is promising for solar energy conversion. Although chemical design and synthesis offers almost unlimited possibility of using new materials and material combinations, the development of efficient bulk heterojunction solar cells requires the optimization of closely interrelated properties at different levels requiring interdisciplinary expertise in various fields of material science.
Acknowledgements
The authors acknowledge the financial support of the European Commission within the framework of Research and Training Network (RTN) project EUROMAP (Contract No. HPRN-CT-2000-00127). A.J.M. thanks Antonio Cravino, Christoph Winder and Harald Hoppe for valuable discussions. Gilles Dennler is acknowledged for his valuable help in preparation of this manuscript. The group of D. Vanderzande is gratefully acknowledged for providing the MDMO-PPV polymers.