

1 Introduction
Since the famous article of Kroto et al. [1] describing the discovery of fullerene C60 20 years ago and its subsequent large scale production at the beginning of the 1990s [2], numerous chemical methodologies have been developed to functionalize C60, in particular for applications in materials science [3]. Its unique electrochemical properties, with six reversible single-electron reduction waves [4], combined with interesting photophysical properties [5], make C60 a fascinating chromophore to study photodriven redox phenomena. Photoinduced electron transfer processes are of great significance in nature since they govern photosynthesis, and considerable efforts are still devoted to constructing bi- or multichromophoric artificial photosynthetic systems, especially those based on donor-bridge-C60 molecules, able to transform sunlight into chemical energy [6,7]. Moreover, the ability of a π-conjugated polymer to efficiently transfer electrons to C60, giving rise to long-lived charge-separated states, has established fullerene derivatives among the most important organic materials for photovoltaic applications [8]. Consequently, during the past 10 years, research programs have been concentrated on elaborating plastic solar cells in which the photoactive layer consists of an interpenetrating network of a π-conjugated polymer acting as the electron donor and a functionalized C60 derivative (PCBM) 1 as the electron acceptor [9]. Based on this concept of a bulk hetero-junction, a power conversion efficiency around 3.5% was reported [10], this latter being very recently improved approaching 5% [11]. Intensive efforts have been recently focused on the development of a molecular strategy to limiting the tendency of C60 to phase separate from the polymer in the polymer/C60 blend. Thus the direct covalent bonding of different π-electron donors [12] and π-conjugated systems [13] has emerged as a very active field of research to reach new organic photovoltaic devices. The utilization of a fulleropyrrolidine derivative attached to an oligophenylenevinylene can be considered as the first example of a molecular hetero-junction specifically designed for photovoltaic conversion [14].
The development of elegant methodologies allowed the attachment of a wide variety of electron donor molecules to the electroactive C60 chromophore. Among these accessible chemical functionalizations, the [4 + 2] Diels–Alder cycloaddition has proved to be one of the most interesting reactions, since the resulting two bonds provide a rigid bridge between donor and C60 to yield systems with well defined geometries. In addition, through–space interactions were expected to be favored in the electron transfer process thanks to a folded-boat conformation of the cyclohexene ring [15]. The versatility and the remarkable selectivity of this Diels–Alder reaction towards the synthesis of fullerene adducts has recently been reviewed [16]. The objective of this account is to present a brief overview of available methodologies for attaching a π-electron donor to C60 through the Diels–Alder reaction, and we focus on the work developed in our group concerning tetrathiafulvalene (TTF) in TTF–C60 dyads, (TTF)n–C60 polyads and C60–TTF–C60 dumbbells.
2 Discussion
2.1 Diels–Alder reaction from endocyclic or acyclic 1,3-dienes and o-quinodimethanes
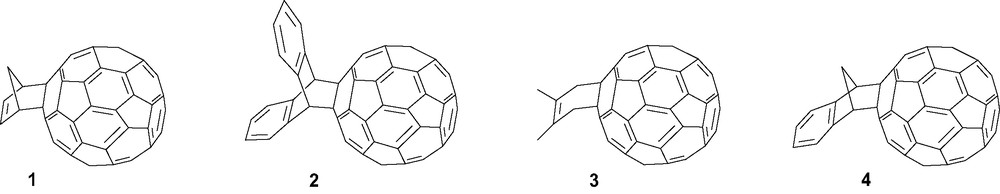
Diels–Alder cycloadditions usually occur between electron deficient dienophiles and electron rich dienes. Therefore, the low LUMO energy of C60 (reactivity equivalent to that of p-benzoquinone) and the dienophilic character of [6,6] double bond facilitate reactions [17]. Among the first reported chemical reactions involving C60 were adducts 1–3, respectively, formed with cycloaddition using cyclopentadiene [18,19], anthracene [17,19,20] and 2,3-dimethylbuta-1,3-diene [21] (Scheme 1). Cycloaddition using isoindene [22], generated in situ from indene, was also reported to yield derivative 4. The synthesis of pentacene-C60 adducts has very recently been reviewed [23].
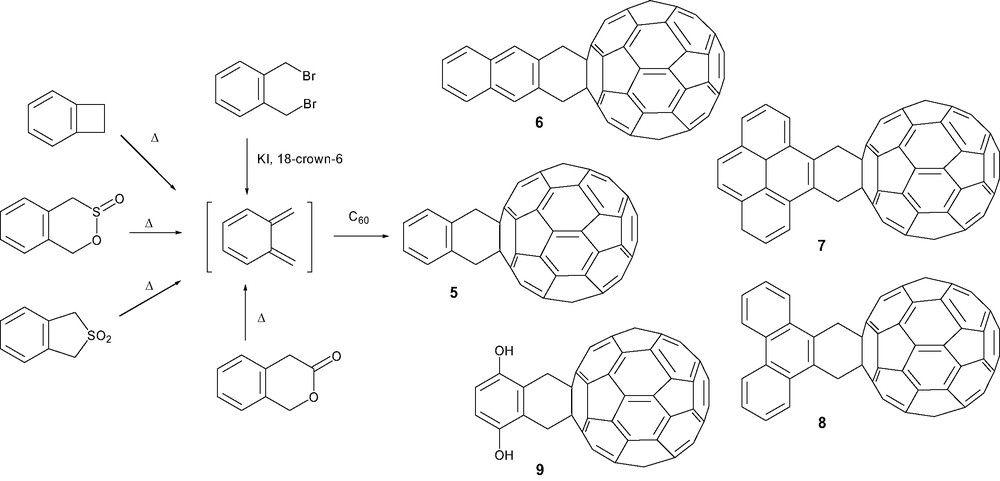
Another approach involving o-quinodimethane derivatives as dienes was introduced by Belik et al. [24] to reach dyad 5 in a method that was extended to naphthalene (6) [25], pyrene (7) [25], phenanthrene (8) [25] and hydroquinone (9) [26] leading to thermally stable cycloadducts. The [4 + 2] cycloaddition required o-quinodimethane precursors [27] generated in situ by various methods such as 1,4-reductive elimination from 1,2-bis(bromomethyl)benzene [24], thermolysis of 1,2-dihydroxycyclobutabenzene [28], thermal extrusion of sulfur dioxide from corresponding sultines [29] or sulfones [30], and extrusion of carbon dioxide from isochromanone derivatives [31] (Scheme 2).
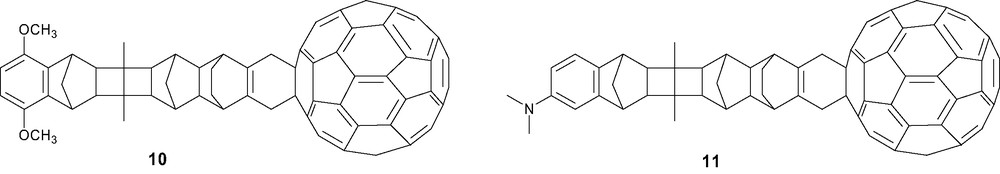
More sophisticated C60-containing donor–acceptor systems with a rigid bridge separating the electron donor group from the fullerene acceptor were constructed. In order to understand how the dynamics of electron-transfer depends on the distance and relative orientation of both redox centers, long-range electron-transfer processes [32] were investigated within covalently linked donor–norbornylogous bridge–C60 systems. ‘Ball-and-chain’ architectures 10 and 11, with 1,4-dimethoxybenzene [33] or N,N-dimethylaniline [34] as the donor-substituted aryl chromophore and a rigid ‘norbornylogous’ hydrocarbon bridge, were synthesized from acyclic 1,3-diene intermediates (Scheme 3). It was clearly showed that rapid long-range intramolecular electron transfer processes can take place in the ‘ball-and-chain’ systems and that the electronic coupling between the C60 chromophore and the hydrocarbon bridge is unusually strong.
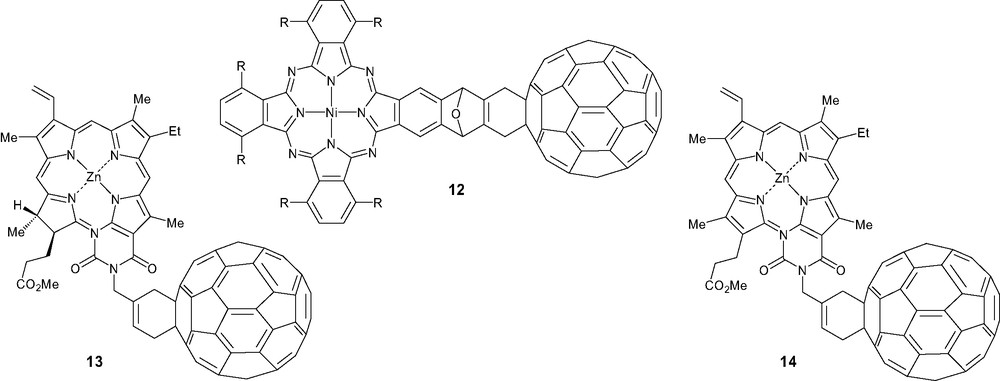
Fullerene C60 and porphyrin have been widely associated as important components for the design of artificial photosynthetic systems in which the porphyrin acts as an antenna for efficient light capture in the visible region of the spectrum. However, it was demonstrated that shortening the separation distance between the donor and the acceptor prolonged the lifetime of the charge-separated state which resulted from the photoinduced single-step electron transfer. Unfortunately, this approach does not seem capable of realizing a high quantum yield of formation of the charge-separated state and it was concluded that a photoinduced multi-step electron transfer strategy would be necessary for the efficient conversion of solar energy into chemical and electrical energies [7]. The Diels–Alder reaction was extensively used in the synthesis of such photoactive systems [6]. Phthalocyanine [35], zinc chlorin and porphyrin [36] were attached to C60 from corresponding acyclic diene intermediates affording systems 12–14, respectively (Scheme 4). The importance of the zinc chlorin chromophore has recently been demonstrated in place of a fully conjugated porphyrin system to obtain long-lived charge-separated state [7].
2.2 Diels–Alder reaction from hetero o-quinodimethanes
Heteroaromatic o-quinodimethanes have received much less attention and appeared to be more difficult to obtain than their non-heterocyclic parents, especially those bearing functionalities that can be altered during the drastic generation process of the intermediate diene. Methods for their preparation [27] included 1,4-reductive elimination of bis(bromomethyl) derivative, thermal treatment of sulfone and cyclobutene derivatives, and flash vacuum pyrolysis (FVP) of benzoate derivatives. Consequently, hetero-Diels–Alder reactions with C60 provided powerful methods to attach for examples thiophene (15) [37,38] and pyrazine (16) [39] (route A), pyrimidine (17) (route B or C) [40], thiazole (18) and oxazole (19) (route B), and also furan (20) (route D) (Scheme 5) [38].
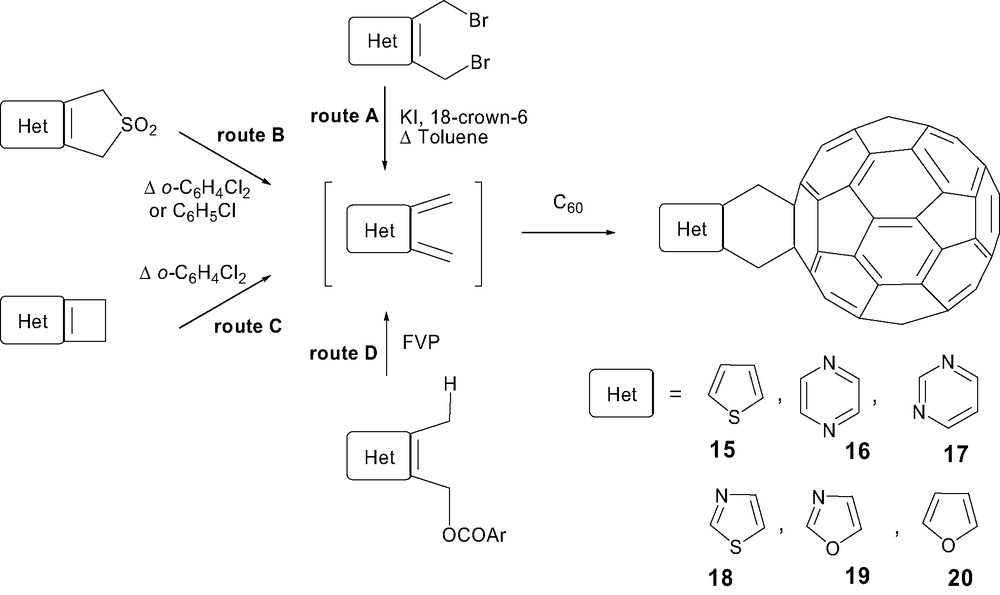
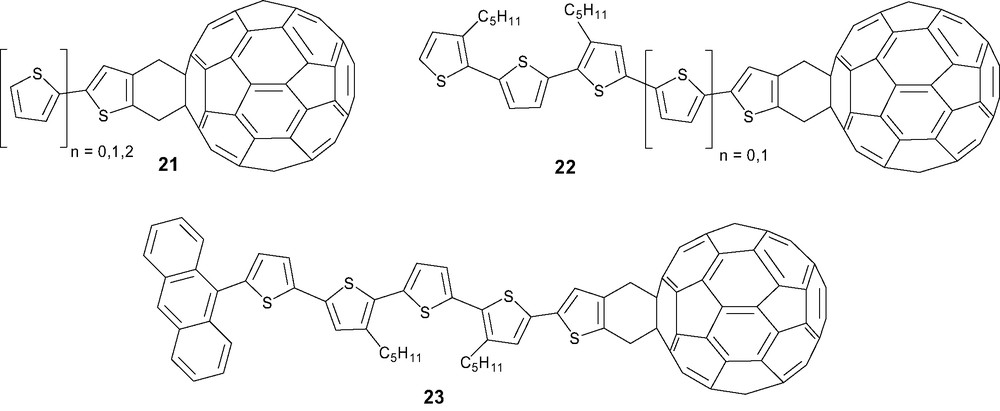
The covalent linkage of an oligothiophene onto C60 was initially investigated by Effenberger et al. Surprisingly, whereas the synthesis of monoadduct 15 in 42% yield started from 2,3-bis(bromomethyl)thiophene, the extension of this strategy to prepare oligothienylfullerenes failed. Corresponding sulfones proved to be more stable precursors and thermal treatment resulted in the generation of the corresponding o-quinodimethane intermediates which were trapped by C60 to afford oligothienylfullerenes 21 and 22 or anthrylthienylfullerenes 23 (Scheme 6) [41].
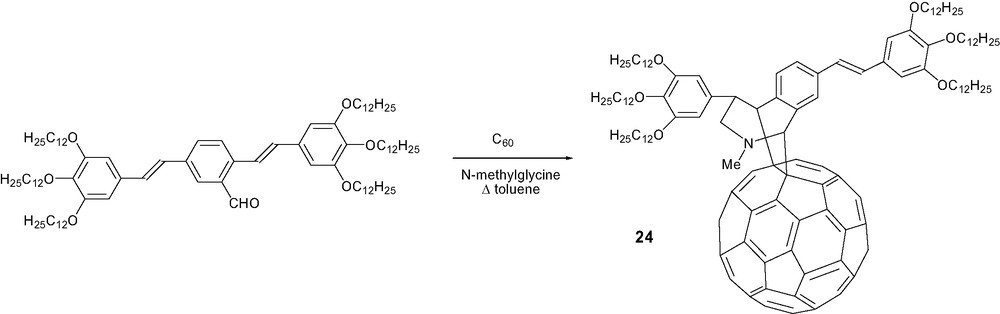
In the search for a molecular approach for organic plastic solar cells, it was first shown that a π-conjugated oligomer, i.e. an oligo(phenylenevinylene) derivative, covalently attached to C60, can be incorporated into photovoltaic devices [14]. As part of this research program, Nierengarten and coll. [42] described dyad 24 as the result of an unexpected Diels–Alder reaction on the fullerene core taking place of the classical 1,3-dipolar cycloaddition (Scheme 7).
2.3 Diels–Alder reaction for linking TTF onto C60
Among the wide variety of donor molecules recently linked to C60, it should be pointed out that most of these donors present an aromatic character in the ground state which is partially or totally lost upon oxidation to yield a π-cation radical species. On these grounds, the strong donor TTF, which is non-aromatic in its ground-state configuration, has emerged as an ideal partner because the stabilization of the charge-separated state TTF+●–C60–● should be enhanced thanks to a gain in aromaticity due to the formation of the thermodynamically stable heteroaromatic 1,3-dithiolium cation upon oxidation [43].
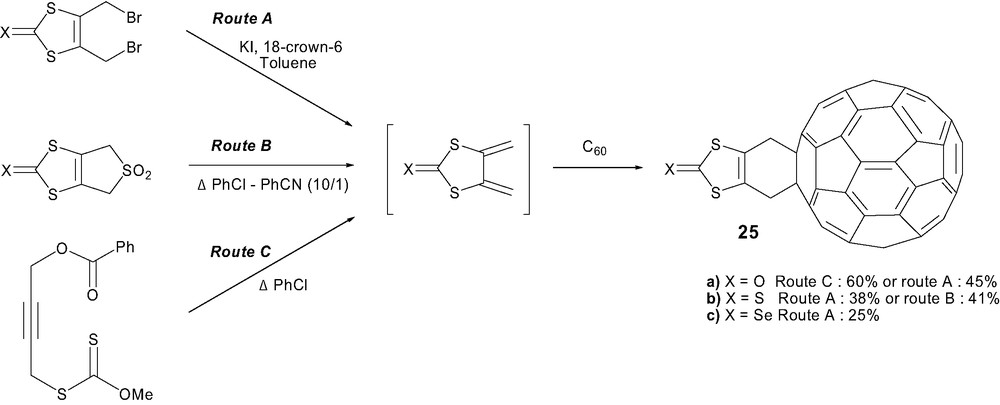
With the aim of attaching TTF onto C60 through a Diels–Alder cycloaddition, groups at Angers [44] and Barcelona [45] independently carried out the reaction in the 1,3-dithiole series. Transient 2-oxo, 2-thioxo or 2-selenoxo-4,5-dimethylidene[2H]-1,3-dithiole intermediates were obtained using different approaches (Scheme 8). The oxo derivative was generated in situ by reductive elimination using naked iodide (route A) [46] or thermal rearrangement of S-propargyl xanthate (route C) [47]. In the thioxo series, classical reductive elimination or thermal treatment of the corresponding sulfone (route B) underwent easy access to the corresponding diene, whereas the selenoxo analog was accessible through route A [48]. Hetero-Diels–Alder reactions with C60 provided efficiently these 1,3-dithiole-C60 dyads 25(a–c) [44,45,49].
Unfortunately, with all attempts to build the TTF core by symmetrical or dissymmetrical coupling reactions being unsuccessful [50], the cycloaddition using the 2,3-dimethylidene[2H]-TTF as the starting material had to be considered. The possibility of developing dienic chemistry in the TTF series was missing until two different strategies were presented by the groups of Angers (route A) [48,51] and Barcelona (route B) [45,52]. Route A used the known detailed synthesis of 2,3-bis(methyloxycarbonyl)TTF. The chemical versatility of ester functionalities was efficiently applied to reach 2,3-bis(hydroxymethyl)TTF and, subsequently, 2,3-bis(bromomethyl)TTF intermediates [53]. For the development of route B, a Wittig-like basic coupling reaction was adapted, promoting the expected dissymmetric TTF starting from both phosphonium and 1,3-dithiolium salts [54]. Reductive elimination using potassium iodide in the presence of 18-crown-6 (route A) or the thermal extrusion of sulfur dioxide (route B) were reactions applied to generate 2,3-dimethylidene[2H]-TTF derivatives. These transient species were trapped by C60 through Diels–Alder cycloadditions providing TTF–C60 systems 26 (Scheme 9). The complementarity of both strategies should be noted when comparing the distinct nature of electron-donating or -withdrawing groups which may be part of the TTF moiety. Various substituents, especially polyalkyloxysulfanyl groups, could be introduced with the aim of increasing solubility properties of dyads for physical applications. It was also possible to replace C60 by PCBM to afford the corresponding dyad 27 in 21% yield as a mixture of regioisomers [55]. Subsequently, route A (the system KI/18-crown-6 was only replaced by Bu4NI) was exploited by Echegoyen and coll. [56] to introduce a crown-ether grafted at the periphery of the TTF core (26h–i). X-ray crystallography on 2-thioxo-1,3-dithiole-C60 adduct 25b showed the half-chair conformation of the cyclohexene ring and the presence of short intramolecular S∙∙∙C distances (3.9–4.0 Å) between sulfur atoms of the dithiole ring and the closest carbon atom of C60 [45]. This value was confirmed by theoretical calculations (4.1 Å) on TTF–C60 dyad 26, as a parameter favorable to efficient intramolecular electron transfer processes.
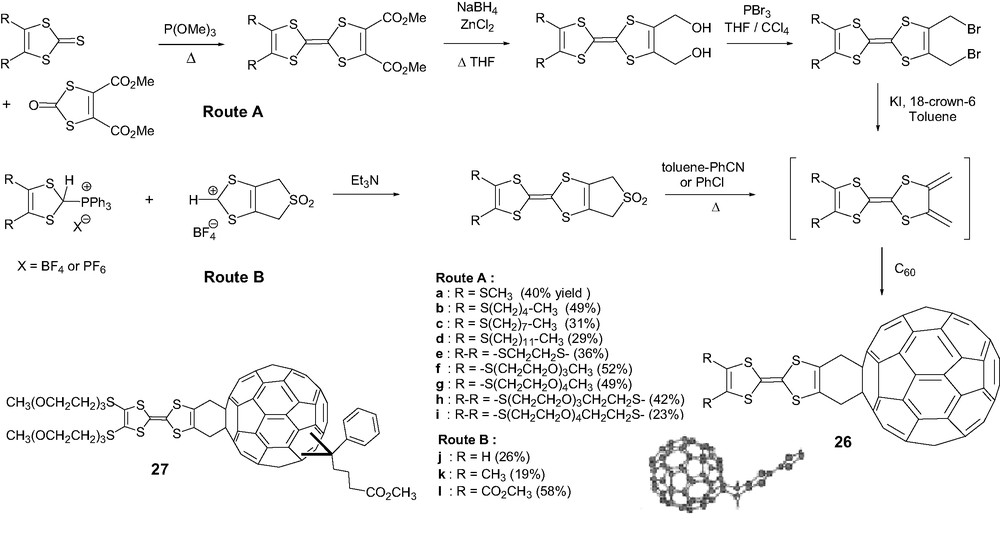
Route A was very recently applied to prepare new TTF–C60 system 28 in which the TTF core was fused with a hydroquinone moiety with tert-butyldiphenylsilyl protecting groups (Scheme 10) [57]. It was demonstrated that their hydrolysis yielded the hydroquinone–TTF–C60 further permitting, due to the presence of hydroxyl groups, the formation of supramolecular structures through a network of hydrogen bonds. The access to the p-benzoquinone–TTF–C60 system 29 after subsequent oxidation of the hydroquinone group is of great interest as it could permit the combination of both charge and electron transfer processes involving TTF with p-benzoquinone [58] and C60, respectively.
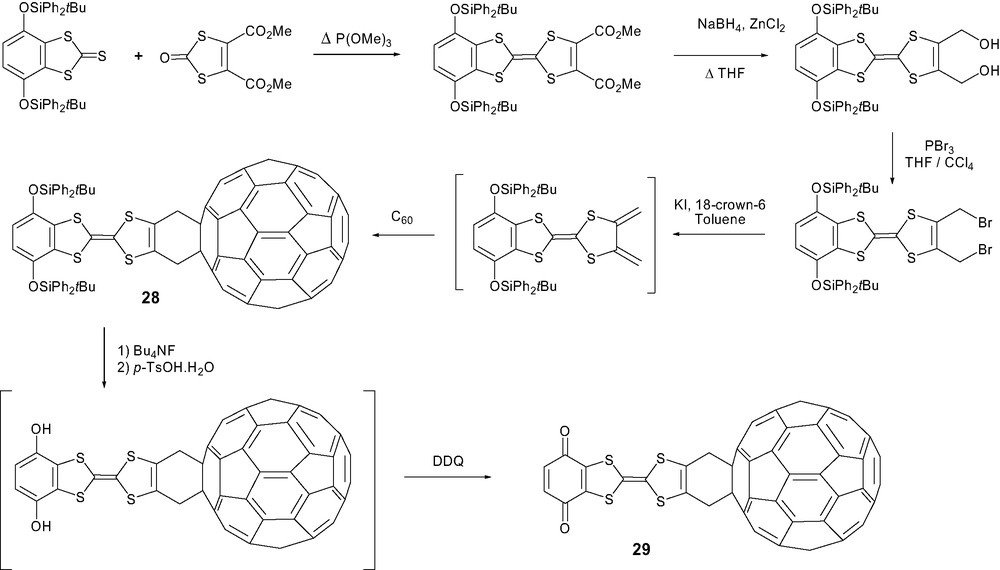
This methodology, starting from 2,3-bis(bromomethyl)TTF derivatives, was applied by Martín and coll. [59] to synthesize the quinonoid π-extended (exTTF–C60) derivative 30 as part of a search for increasing lifetimes of charge-separated states (Scheme 11). Whereas the TTF–C60 dyads yielded radical pairs having lifetimes of a few nanoseconds, the corresponding exTTF–C60 dyads have lifetimes in the range of several hundred nanoseconds. It should be added that the impact of a second exTTF unit was clearly established for an exTTF1–exTTF2–C60 triad presenting exceptional lifetimes of 111 μs in DMF [60].
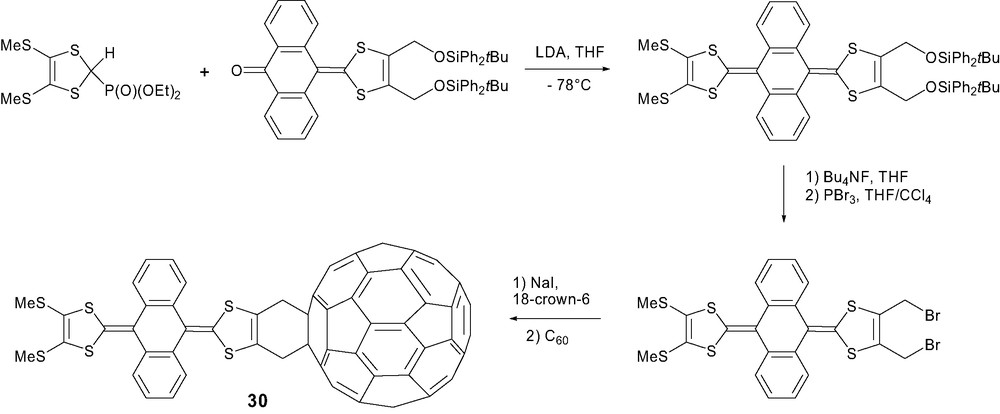
2.4 Multiple cycloadditions of TTF onto C60
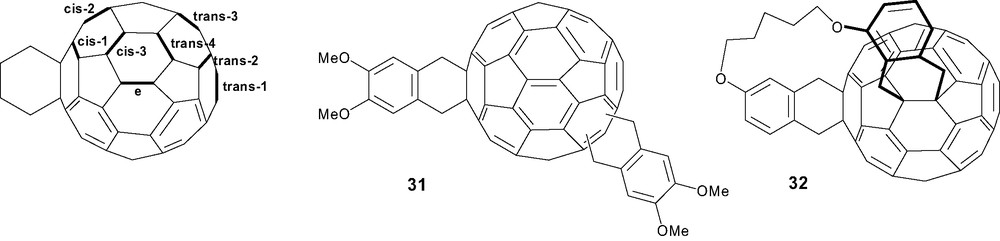
Only a few examples of multiple adducts of [60]fullerene have been investigated due to the problems of isolation and characterization in relation to the number of possible regioisomers which drastically increase with the number of additions. In the bisaddition (Scheme 12), trisaddition and tetrakisaddition of symmetrical addends occurring exclusively at [6,6] junctions, respectively, 8, 46 and 262 regioisomers are theoretically expected [61]. The Diels–Alder reaction of the o-quinodimethane intermediate generated from 1,2-bis(bromomethyl)-4,5-dimethoxybenzene was studied in the case of bisaddition and seven regioisomers of compound 31 were isolated and fully characterized. The regioselectivity differs to that observed for the Bingel-type reaction with a preference for the trans-3 rather than the equatorial regioisomer, and an absence of cis-1 and low yields of cis-2 and cis-3 regioisomers due to the steric repulsion between both cyclohexene rings [62]. In order to limit the number of regioisomers, synthetic methodologies were efficiently developed for controlling multiple additions using tethers as covalent templates [63]. Bis(o-quinodimethane) precursors were prepared and the resulting regioselectivity depends on the tether length. The clear definition of the linkage allowed both cycloadditions to occur selectively within one hemisphere of C60 and the formation of the trans isomer was excluded. Only the equatorial regioisomer 32 was isolated while low amounts of other isomers were detected (Scheme 12) [64].
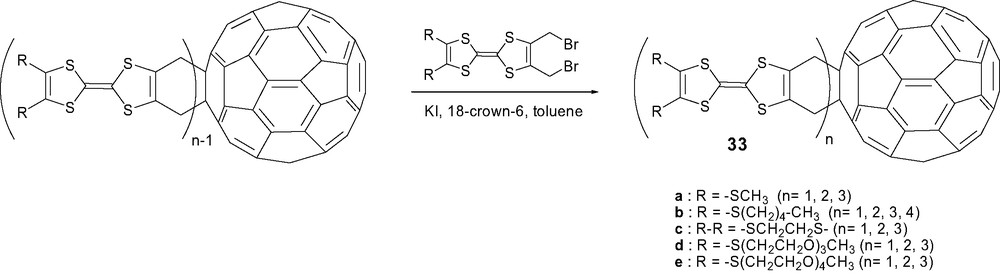
With the aim of limiting charge recombination, which decreases the lifetimes of charge-separated states, the sequential assembly of several electroactive units onto C60 giving rise to triads, tetrads, pentads was considered as a possible contribution to larger lifetimes due to the cascade of charge transfer steps. An additional interest was to isolate new bisadducts and unprecedented tri and tetrakisadducts thus providing access to three-dimensional functionalized fullerenes incorporating several electroactive units. These polyads 33 were formed in very low yields by treating 2,3-bis(bromomethyl)TTF with naked iodide in the presence of C60 according to Scheme 9. More interestingly, their synthesis was considered in higher yields using an iterative strategy by successive treatments of the major adducts produced at each step (Scheme 13) [65]. Nevertheless, the tedious isolation and characterization of the different regioisomers has not been carried out for polyads 33. This difficult work of separation and assignment of the correct structure of regioisomers was only achieved by Rovira and coll. [66] with the triad C60–(TTF)2 in which the TTF bears methoxycarbonyl groups (R = CO2Me).
2.5 Diels–Alder cycloaddition for the synthesis of dumbbells
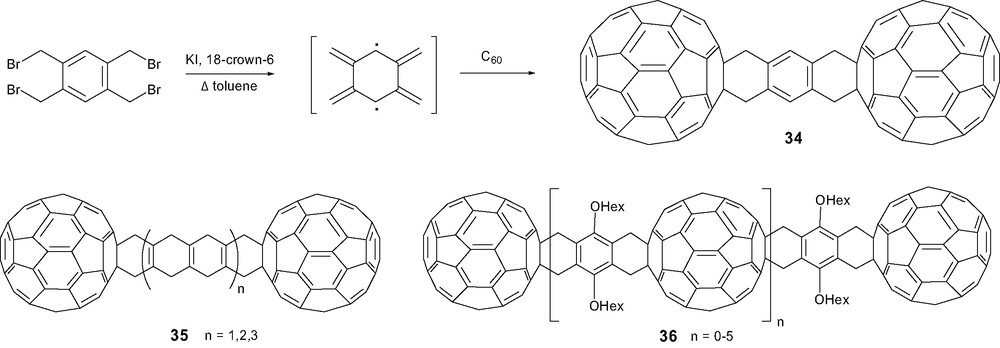
The reaction of bis-o-quinodimethane, prepared in situ from 1,2,4,5-tetrakis(bromomethyl)benzene, with C60 was certainly the first described bridged dumbbell 34. This methodology was extended to the synthesis of dumbbell-like structures 35(a–c) reported as very insoluble compounds and consequently not fully characterized (Scheme 14) [67]. When a similar strategy was used starting from the bis-sulfone as the bis-diene precursor, the resulting oligomer 36(a–e) advantageously displayed advantage of exhibiting reasonable degree of solubility due to the presence of hexyloxy groups [68].
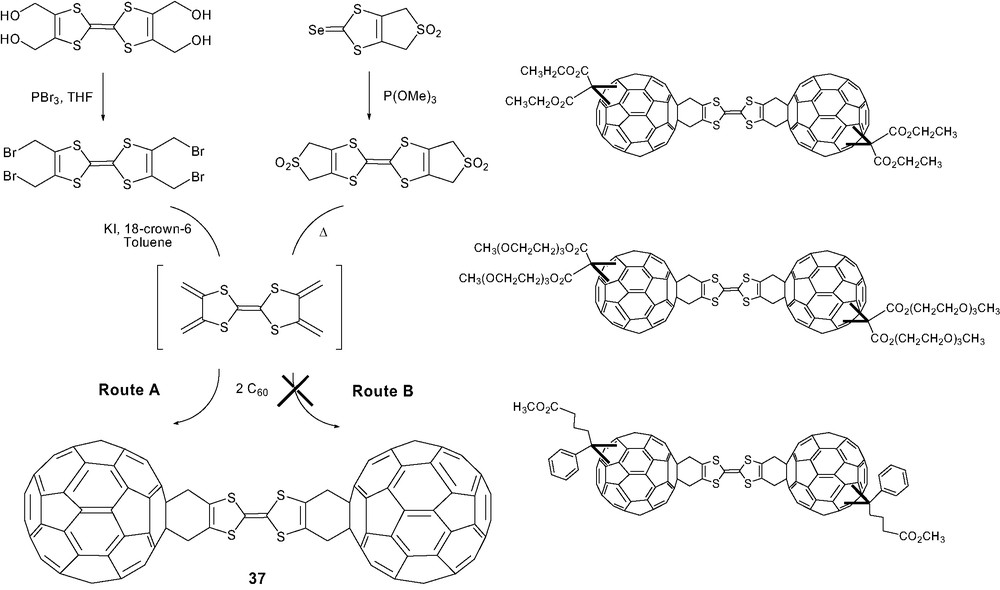
Not long after, we were interested in the synthesis of the C60–TTF–C60 dumbbell 37 [51]. However, this research area was limited by lack of suitable TTF building blocks able to generate tetramethylidene[4H]–TTF. A previous contribution was related to the synthesis of symmetrical TTF bearing two sulfone groups [69]. Nevertheless, no evidence for the formation of cycloadducts from tetramethylidene[4H]-TTF with C60 was observed probably due to the drastic experimental conditions responsible for the polymerization of bis-diene. The isolation of the tetrakis(bromomethyl)TTF [70] and its transformation under milder conditions by reduction elimination constituted key steps in the synthesis of the dumbbell 37 (Scheme 15). Despite its poor solubility, the structure of dumbbell 37 was unambiguously identified thanks to complementary electrochemical and spectroscopic techniques [71]. In order to perform further photophysical studies on such assemblies [72], we developed the synthesis of analogous dumbbells replacing C60 by the methanofullerene derivatives substituted with solubilizing groups [48]. C60-based dumbbells were expected to present highly stabilized charge-separated states but the contribution and the beneficial effect of the second fullerene unit remain unclear up till now [73].
3 Conclusion
One century ago after the reaction between cyclopentadiene and quinones was carried out by Albrecht [74] in 1906, who unfortunately assigned erroneous formulas to the addition products, and the first reported Diels–Alder [75] reaction in 1928, the [4 + 2] cycloaddition is still one of the most important tools used by organic chemists to construct new molecular systems. Dienophilic properties of the fascinating and still recently discovered C60 have been widely exploited in such cycloaddition reactions. This approach appears to be particularly attractive and has been fruitful in preparing rigidified donor–bridge–C60 electroactive systems for various applications in materials science. In particular, straightforward syntheses of 2-oxo (or thioxo or selenoxo)-1,3-dithiole–C60 systems, TTF–C60 dyads, (TTF)n–C60 polyads and dumbbell C60–TTF–C60 were carried out using [4 + 2] Diels–Alder cycloadditions. The key step consisted in the preparation of versatile bis or tetrakis(bromomethylated) derivatives which could generate corresponding transient o-quinodimethane heteroanalogues via reductive elimination under relatively mild synthetic conditions. The advantage of the Diels–Alder approach concerns the more rigid spatial orientation of the HOMO of the donor with respect to the LUMO of C60. Moreover, the introduction of different functional groups on the TTF core allows a tuning of the energy gap between the frontier orbitals of the dyad [23]. Consequently, the possibility of modulating the electron transfer process is attractive in designing new artificial photosynthetic models and electronic devices. It has also been demonstrated using the 1,3-dithiole and TTF series that applications outside of those expected are possible due to their exhibiting nonlinear optical and optical limiting properties [76].
Acknowledgments
I would like to warmly thank all PhD students, post-doc researchers, co-workers and colleagues from Angers, whose names appear in the references, for their outstanding contribution to this research topic. I particularly thank Dr. C. Rovira and Professor J. Veciana (ICMAB, Barcelona) for their fruitful collaboration. Dr. R.C. Hiorns is thanked for his help in the preparation of this manuscript.
1 PCBM = methano [60]fullerene [6,6]-phenyl C61 butyric acid methyl ester.
Vous devez vous connecter pour continuer.
S'authentifier