

1 Introduction
Bismuth, based oxide materials are receiving considerable attention for their numerous potential applications including as oxidation catalysts [1], next generation memory devices [2], high Tc superconductors [3], high temperature electrolytes [4] and nonlinear optical materials [5]. Additionally, bismuth oxide has been used for the oxidative coupling of methane and for C–H bond activation and radical-surface reactions for propylene and methane [6]. Additionally, some bismuth vanadates are photocatalysts whose activity has been described as being more than five times that observed for TiO2 for the decomposition of water [7]. Many binary bismuth oxides are thermochromic and are being explored as environmentally benign alternatives to replace cadmium or lead based systems [8].
Arguably, one of the most active areas of research in bismuth oxides centers on the investigation of new ferroelectric materials and their use in the development of new nonvolatile random access memories (NVRAM). Of particular significance are a series of layered oxide materials known as the Aurivillius phases. All of the Aurivillius phases show significant ferroelectric behavior and are, therefore, potential candidates for ferroelectric random access memories (FRAM). The composition of the Aurivillius phases can be expressed by the general formula (Bi2O2)2+(An–1BnO3n+1)2– where A and B can be any metal ion with the appropriate charge to radius ratio [9]. Most commonly, A is Bi, Pb, Sr, Ca or K and B is Ti, Zr, Nb, Ta, Mo, W, Fe or Co. Of all the potential combinations that have been created from the general formula, those containing the transition metals titanium, niobium or tantalum have shown the greatest promise for the development of significant ferroelectric materials [10,11].
The performance of many bismuth-based oxide systems is sensitive to the synthesis conditions [12]. Oxides produced under mild conditions, such as sol–gel [13], metal organic decomposition (MOD) [14], chemical bath deposition [12], ionic layer deposition [15,16], or co-precipitation techniques [17,18], often display improved properties compared to that achieved from traditional solid-state syntheses. It has recently been shown that the use of molecular single-source precursors often allows for the formation of crystalline bimetallic or multimetallic oxides at conditions even milder than what can be achieved by sol–gel or co-precipitation routes [19]. This has been attributed to the intimate and controlled mixing of the metal species at the molecular level, which greatly facilitates the crystallization process of the product oxide [20]. Furthermore, these precursors can be used with both spin-casting techniques to form oxide thin films or with thermolytic or hydrolytic techniques to form discrete crystallites. Consequently, we have sought to develop a rational approach to the synthesis of bismuth-transition metal oxides through the use of discrete well-defined molecular precursors. We have further sought, where possible, to ensure that the synthetic approaches developed were flexible and general enough to allow for the inclusion of a wide range of transition metal species so that the effects of doping on the chemical and physical properties of the product oxides might be investigated directly.
However, the synthesis of such heterobimetallic coordination compounds remains complicated by the frequently divergent electronic and coordination requirements of the two or more different metal species present in the target complex. These problems are further exacerbated when attempting to form discrete molecular complexes involving bismuth, due to the fact that this metal is an extremely strong Lewis acid and can readily expand its coordination sphere to form hypervalent coordination complexes. The result of these physical characteristics is that many simple bismuth coordination complexes tend to be intractable coordination polymers or oligomers. Additionally, bismuth rarely adopts a regular coordination geometry, which makes the ‘rational’ design and synthesis of heterobimetallic species still more problematic.
As a result of these features, there is relatively little structural information available for bimetallic alkoxide or carboxylate complexes of bismuth and other metals. Known compounds are limited to several alkali or alkaline-earth metal species [10,21,22]. A few bismuth–transition metal alkoxide complexes such as [BiCl3OV(OC2H4OMe)3]2 [23], BiTi2(μ3-O)(μ-OiPr)4(OiPr)5 [24] Cp2MoBi(μ2-OEt)2(OEt)2Cl (Cp = C5H5) [25], have been reported. Bismuth–molybdenum allyl carbonyl complexes such as [(μ3-CHC(CH3)CH2)Mo(CO)2(μ2-OEt)3Bi(OEt)]2 have been characterized [26]. Also, two substituted polyoxometalates of general composition [NBu4]3[M4Mo(μ5-O)2(μ2-OMe)4(μ2-O)8(NO)(O)4]2Bi (M = Mo, W) are known [27]. Of the compounds reported, [BiCl3OV(OC2H4OMe)3]2, Cp2MoBi(μ2-OEt)2(OEt)2Cl and many of the allyl–molybdenum complexes contain halide ions that would likely act as a contaminant of the final oxide material. The polyoxometalates do not contain the correct stoichiometry of metals to make them useful precursors for the formation of the ferroelectric Aurivillius phases. This leaves only BiTi2(μ3-O)(μ-OiPr)4(OiPr)5, Bi4Ba4(μ6-O)2(μ3-OEt)8(μ-OEt)4(η2-thd)4 (thd = 2,2,6,6-tetramethylheptanedione), Mo2Bi2(CO)4(μ2-OCH2CH3)8(η3-CH2C(CH3)CH2)2 and possibly [MoBi(CO)2(μ2-OCH2CH2OCH3)3(η3-CH2C(CH3)CH2)(THF)][BF4] as well-characterized potential precursors for advanced oxide materials based on bismuth [28].
We report in this account recent progress in our laboratory on the preparation and reactivity of heterobimetallic bismuth based coordination complexes involving both early (e.g. Ti, Nb and Ta) and late (e.g. Cu, Ni and Co) transition elements. Our synthetic approach relied on the utilization of the bifunctional ligands derived from salicylic acid (Fig. 1: Hsal–, HOC6H4CO2–; sal2–, OC6H4CO22–) to direct the stepwise introduction of the different metal species into the final compound for the early transition metals, as illustrated in part 1 of Fig. 2-1. Alternately, in the case of the late transition elements, we were able to use pre-formed metal complexes M(acac)3 (acac = acetylacetonate) and M(salen) (salen = ethylenebis-salicylimine) as ligands to coordinate the bismuth center through direct Lewis acid–base adduct formation (Fig. 2-2). We discuss here these two versatile routes to the formation of discrete, molecular heterobimetallic complexes of bismuth and the utility of these compounds in the formation of advanced materials, including both bulk oxides and discrete nanoparticles.

Table of potential coordination modes of salicylic acid between one or more metal centers.

Detail of the general synthetic strategies employed for the production of heterobimetallic coordination complexes of bismuth salicylate via either a bifunctional ligand (1) or Lewis acid–base adduct (2) approach.
2 Heterobimetallic complexes of bismuth(III) salicylate
2.1 Bi–Ti heterobimetallic complexes
Despite the extensive use of salicylate salts of bismuth in pharmaceutical applications [29], very little is known of the reaction chemistry of this material, particularly with respect to the interaction this complex with other metal species. We found that it is possible to prepare a soluble, and consequently reactive, form of bismuth saliyclate through the acidolysis of triphenylbismuth with salicylic acid in refluxing toluene [28]. The salicylate ligand shows considerable diversity in its metal binding properties from its ability to act as Hsal– or sal2–, as well as utilizing both the carboxylate and phenolic functions (Fig. 1). While the structure of the [Bi(Hsal)3]n has not been conclusively determined, we have found that it is a versatile reagent for the formation of heterobimetallic coordination complexes through reaction with the metal alkoxide Ti(OiPr)4 via alcohol elimination reactions [30]. Considerable diversity in the structural and chemical properties of the resulting heterobimetallic species can be obtained through adjustment of the reaction conditions including solvent polarity, water concentration, concentration of salicylic acid and metal stoichiometry, among others. Consequently, we have been able to employ this synthetic approach to produce numerous heterobimetallic species including Bi2Ti3(sal)8(Hsal)2, Bi2Ti4(sal)10(Hsal)(OiPr)∙H2O, BiTi4(sal)7(OiPr)6, Bi4Ti4(sal)10(OiPr)8 and Bi8Ti8(sal)20(OiPr)16 [30]. A diagram of the solid-state structure of BiTi4(sal)6(OiPr)7 [30b], which provides a general representation of the nature of interaction between the bismuth carboxylate and the transition metal alkoxide, is provided in Fig. 3.
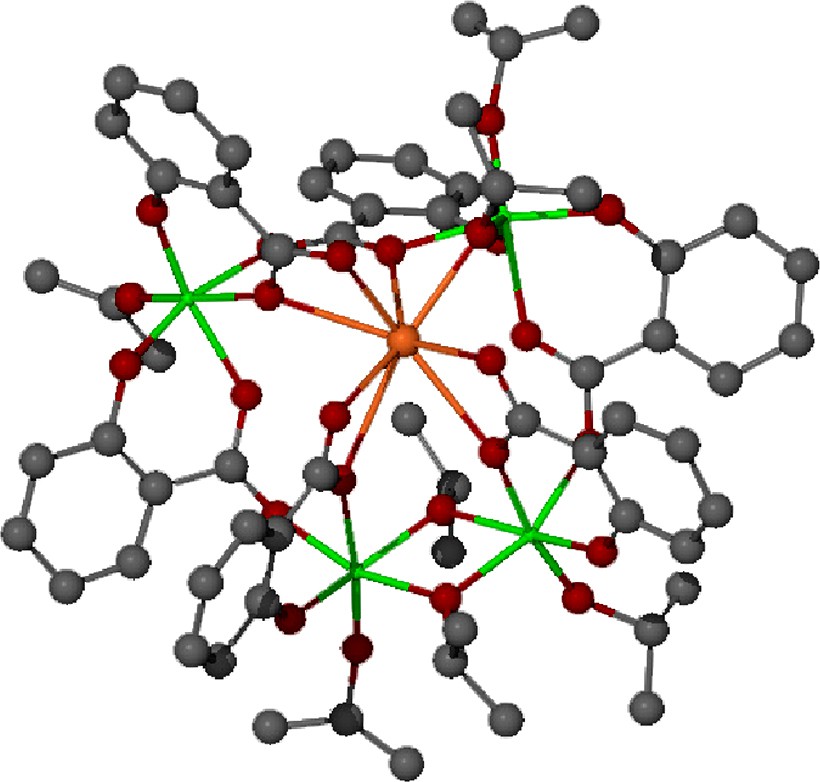
Ball-and-stick representation of the solid-state structure of BiTi4(sal)6(OiPr)7. In the illustration, bismuth is orange and the transition metal is green [30b].
The driving force for the formation of the heterobimetallic species using this synthetic approach appears to lie in the acidity of the residual phenolic protons in [Bi(Hsal)3]n, coupled with the ability of the salicylate ligand to form a stable chelate complex with the transition metal center. It is important to emphasize that several of these complexes were observed to co-crystallize with water, illustrating that the salicylate ligand is not only able to serve as an effective scaffold for orientation and association of the two metal species with one another, but the interaction of the salicylate ligand with the transition metal center is able to appreciably attenuate the hydrolytic reactivity of the residual alkoxide ligands and prevent premature hydrolysis. Indeed, several of the compounds described here can only be isolated if small amounts of water are present in the reaction mixture. Multinuclear NMR experiments [30b] of these compounds give results that are consistent with the structural and symmetry considerations derived from the crystallographic data, thereby suggesting that these compounds are able to retain their solid-state structure in solution. This structural stability coupled with the stability of these complexes against unwanted hydrolysis suggests that these systems may be viable precursors for the formation of advanced oxide materials by solution-based routes. This resistance to unwanted hydrolysis coupled with spectroscopic evidence that many of these complexes are able to retain their solid-state structure in solution, suggests that the compounds Bi4Ti4(sal)10(OiPr)8 and [Bi4Ti4(sal)10(OiPr)8]2 were both isolated from a mixture of bismuth salicylate and titanium isoproxide that was rich in the transition metal species. These complexes are structurally important as they represent examples of heterobimetallic coordination complexes that are equimolar in both a heavy main group element and a transition metal. The difficulty in preparing such complexes has been previously observed, and has been attributed to the higher coordination requirements of the main group metal [31]. As a result of these requirements, heterobimetallic species almost invariably are rich in the transition element. Here, it appears that the chelating ability of the salicylate ligand, both through the carboxylate and phenolic oxygen atoms as well as through η6 interactions of the arene ring, is able to control the coordination environment of the bismuth to an extent that the isolation of 1:1 complexes is possible.
This work illustrates that bismuth salicylate is a versatile reagent for the formation of heterobimetallic coordination complexes. Initially, it was assumed that the likely product of these reactions would be the formation of complexes in which the metal centers were assembled by bridging interactions between carboxylate and oxo ligands. This chemistry has wide precedent in the interaction of other main group metal carboxylates (principally acetates) with titanium alkoxides. However, the formation of oxo or hydroxo ligands was not observed in any of the reactions that we have attempted. This is likely due to the stability of the salicylate chelate interaction with the metal center. It appears, in this case that the Lewis acidic nature of the bismuth atoms is satisfied by bridging interactions with the titanium centers, and by interaction with the arene ring of two of the salicylate ligands. The impetus for the ligand transfer is likely tied to the strong Lewis basicity of the alkoxide ligand and to the stability of the O,O′ chelate that the salicylate ligand is able to form with the titanium center.
2.2 Bi–Nb and Bi–Ta heterobimetallic complexes
Like titanium, the alkoxides of niobium and tantalum readily react with bismuth salicylate to produce heterobimetallic coordination complexes under very mild conditions. The complexes produced from the reaction of niobium or tantalum alkoxides with bismuth salicylate and salicylic acid can be generally grouped into two categories: one containing complexes in which the metal centers interact only through bridging salicylate ligands (i.e. Bi2M2(sal)4(Hsal)4(OiPr)4 (Fig. 4) and Bi2M2(sal)4(Hsal)4(OEt)4 (M = Nb, Ta)) and another in which the transition metal centers are assembled through μ-oxo ligands (Bi2M2(μ-O)(sal)4(Hsal)4(OEt)2·H2O and BiM4(μ-O)4(Hsal)3(sal)4(OiPr)4 (M = Nb, Ta)) [30a]. As with the titanium-based complexes, there is a complex dependence on the solvent system employed in the reaction, the concentration of water in the reaction mixture and, potentially on the pH, to determine whether one or more oxo ligands will be produced. The solution-state behavior of these complexes appears to vary dramatically from that of the titanium-based systems.

Ball-and-stick representation of the solid-state structure of Bi2Nb2(Hsal)4(sal)2(OEt)2(μ-O) [30a]. In the illustration, bismuth is orange and the transition metal is green.
Heterobimetallic complexes containing bismuth and niobium or tantalum can be readily synthesized in a stepwise manner if careful control is paid to the nature of the organic ligand that is employed in the synthesis and to the reaction conditions that are used in the formation of the product. The products display influences of the coordinative flexibility of the salicylate ligands, the relative solubility of the bimetallic products in a given solvent system, the oxidation state of the transition metal, the tendency of the transition metal to remain octahedral and the ability of bismuth to easily expand its coordination sphere. It seems likely that the cumulative effects of these traits, acting in concert, are what ultimately determine the composition and structure of the bimetallic complexes.
2.3 Lewis acid–base adducts of bismuth salicylate
While the reaction of bismuth salicylate [Bi(Hsal)3]n with transition metal alkoxides produces stable heterobimetallic species under very mild conditions and in appreciable yields, direct stoichiometric control over the relative ratios of the two metal species in the final products have been difficult to achieve using this synthetic approach. The ability to ‘design’ such complexes remains elusive. As a result, we have explored alternate routes to the formation of heterobimetallic coordination complexes of bismuth. β-Diketonate complexes of the general form M(acac)3 have been shown to form stable complexes with a broad range of the transition and main group elements and the lanthanides. Consequently, we have investigated the ability of these systems to form Lewis acid–base adducts with bismuth(III) salicylate, with the intention of producing a general route to the formation of heterobimetallic coordination complexes for potential use as precursors for advanced oxide materials.
As noted in the introduction, bismuth salicylate possesses a significant Lewis acidity that we have been able to exploit for the synthesis and isolation of the complexes [Bi(Hsal)3(bipyridine)]2·2 toluene and [Bi(Hsal)(sal)(1,10-phenanthroline)]2·2 toluene [32]. Similarly, we have found that bismuth(III) salicylate reacts readily with trivalent metal diketonate complexes to form 2:1 adducts of the formula Bi2(Hsal)6·M(acac)3 (M = V, Al, Fe, Co, Cr) [33]. The solid-state structure of Bi2(Hsal)6·Al(acac)3 is illustrated in Fig. 5.
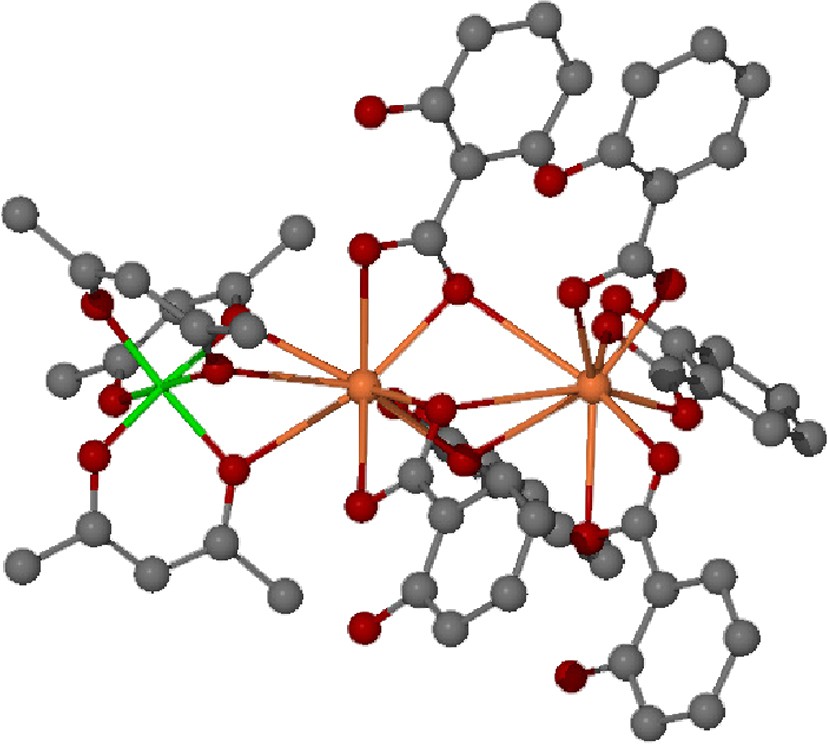
Ball-and-stick representation of the solid-state structure of Bi2(Hsal)6∙Al(acac)3 [30a]. In the illustration, bismuth is orange and aluminum is green.
Despite the strong structural similarities between the complexes, there is a dramatic difference in stability of the solutions of Bi2(Hsal)6·Al(acac)3 and Bi2(Hsal)6·Co(acac)3. Unlike Bi2(Hsal)6·Al(acac)3, which appears to be stable for several days in solution, Bi2(Hsal)6·Co(acac)3 undergoes rapid dissociation on dissolution, so that the only species that is detected by 1H NMR is Bi(Hsal)3·Co(acac)3. In this case, the aromatic region of the spectrum shows the anticipated through-bond coupling consistent with the presence of a single ligand environment [33]. The reasons for the difference in stability of solutions of Bi2(Hsal)6·Al(acac)3 and Bi2(Hsal)6·Co(acac)3 are not clear.
As with the alkoxide derived heterobimetallic species, multinuclear (13C and 27Al) NMR experiments [33] suggest that, with the exception of M = Co, these species are able to dissolve into in non-coordinating solvent intact. In the case of the cobalt complex, dissolution results in rapid elimination of one of the “Bi(Hsal)3” units, so that Bi(Hsal)3·Co(acac)3 is the only species that is detected. Direct evidence for interaction of the β-diketonate complex with the bismuth carboxylate has been obtained from the 27Al MQ-MAS NMR spectrum of Bi2(Hsal)6·Al(acac)3, which shows a single resonance with peaks at δ = 1.59 and –0.18 ppm (Fig. 6a). This chemical shift of this peak represents a significant downfield shift versus free Al(acac)3 (δ = –1.57 and –4.72 ppm), and is consistent with the electronically deshielding nature of bismuth. Similar effects have been observed in other bismuth coordination complexes including alkoxides [24] and diketonates [10]. Similarly, the solution-state 27Al NMR spectrum displays a single resonance at δ = 0.88 ppm, which is shifted from that of free Al(acac)3 (δ = 0.66 ppm) (Fig. 6b).
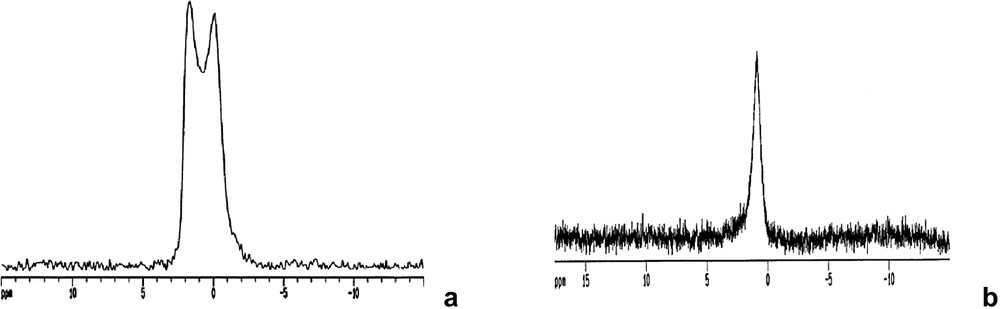
(a) MAS 27Al NMR spectrum of Bi2(Hsal)6.Al(acac)3, 20-kHz MAS was employed during data collection. The experiment consisted of 0.3us pulse (using 1-kW amplifier, solution 90 pulse = 0.97, 41 ms FID, and 2-s relaxation delay. 256 scan were collected, no line broadening was employed in data processing. (b) Solution-state 27Al NMR spectrum of Bi2(Hsal)6·Al(acac)3 shows a δ = 0.88 ppm confirming the interaction between the two centers. (Used by permission of the American Chemical Society).
The formation of the complexes Bi2(Hsal)6·M(acac)3 (M = Al, Cr, Fe, V, Co) may illuminate the nature of [Bi(Hsal)3]n in solution. Since [Bi(Hsal)3]n is not very soluble once isolated, it is reasonable to assume that it tends towards oligomerization, and that oligomerization may occur in a fashion similar to the way the two Bi(Hsal)3 groups are nested together in the M(acac)3 adducts [30]. The precipitation of the starting material can be avoided if the compound is prepared carefully and re-dissolved in methanol immediately. Upon dissolution, the species may still be made up largely of oligomers, and it may be likely that n = 2 in this solution-based upon the stoichiometry in the resulting adducts. Thus, addition of M(acac)3 (M = Al, Cr, Fe, V, Co) would proceed immediately to produce the kinetically favored insoluble [Bi(Hsal)3]2·M(acac)3 (M = Al, Cr, Fe, V, Co), as has been confirmed by single crystal X-ray diffraction for the Al compound. These products form even in the presence of excess M(acac)3 (M = Al, Cr, Fe, V, Co) suggesting that the rearrangement [Bi(Hsal)3]2·M(acac)3 + M(acac)3 → 2 Bi(Hsal)3·M(acac)3 does not have time to occur in solution before precipitation takes place.
While the use of the diketonate complexes proved to be an extremely general method for the formation of heterobimetallic compounds of bismuth in which the second metal was trivalent, we have not yet been successful in extending this chemistry to include divalent transition metal species (e.g., Cu(acac)2, Ni(acac)2). In contrast, we have found that the use of the salen and salen* ligands (salen = bis(salicyl)imine; salen* = bis(3-methoxysalicyl)imine) in place of the diketonate allows for the isolation of heterobimetallic species including Bi(Hsal)3∙Cu(salen) (Fig. 7), Bi(Hsal)3∙Ni(salen), Bi(Hsal)3(sal)∙VO(salen*) and Bi2(Hsal)6∙Co2(salen)3. All of the species that have been structurally characterized [33], including the copper, nickel and cobalt species, interact with the bismuth center through the phenolic oxygen atoms of the salen ligand. The steric constraints of the salen ligands appear to result in the formation of the 1:1 heterobimetallic adducts exclusively.
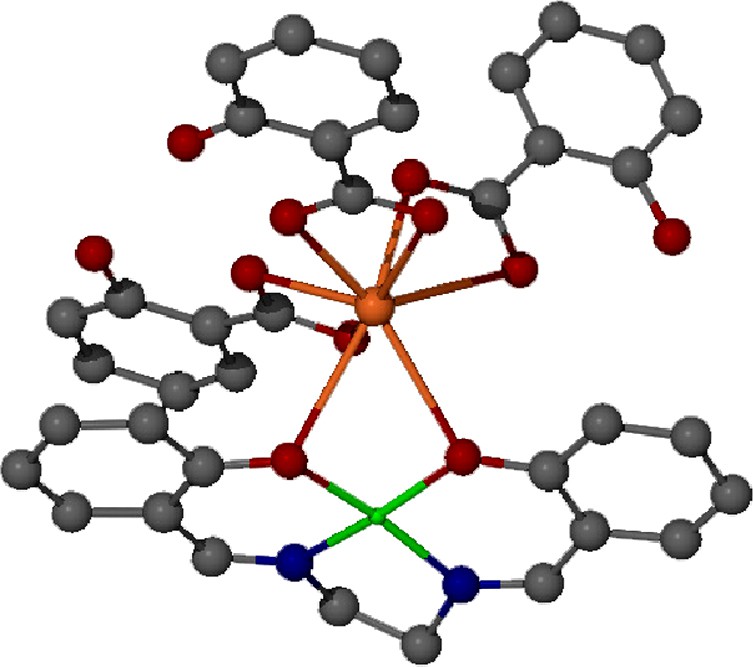
Ball-and-stick representation of the solid-state structure of Bi (Hsal)3∙Cu(salen). In the illustration, bismuth is orange and copper is green [45].
3 Utility of heterobimetallic bismuth coordination complexes as single-source molecular precursors for advanced oxide materials
3.1 Thermal decomposition studies of titanium alkoxide derived complexes
We have investigated the thermal or hydrolytic decomposition of several of the complexes presented previously to ascertain both the phases that are produced, the morphology of the final oxide and the minimum sintering temperature required to produce crystalline nanomaterials. The thermal decomposition of the complexes has been investigated by simultaneous thermogravimetric analysis/differential thermal analysis (TGA/DTA) to monitor phase changes and the onset of crystallization in the materials, as well as to exclude the possibility of any deleterious processes such as sublimation events during the thermal decomposition of the molecular precursor. The morphology and composition of the materials produced in this manner has been probed by scanning electron microscopy (SEM) and powder X-ray diffraction.
The thermal decomposition of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O and Bi2Ti3(sal)8(Hsal)2 were investigated by thermogravimetric analysis and by powder X-ray diffraction. The thermal decomposition of Bi2Ti3(sal)8(Hsal)2·3CH2Cl2 proceeds with two distinct losses of mass are observed on pyrolysis of the sample. The initial step, which occurs between 37 and 152 °C and represents a loss of 12% of the sample, corresponds to the loss of lattice solvent from the sample (Fig. 8-1). The observed mass loss during this step is in exact agreement with the loss calculated for the removal of three molecules of dichloromethane from the sample (ca. 12%), as predicted by the X-ray diffraction data. Decomposition of the complex occurs in a single step beginning at 278 °C and reflects a 64% loss of mass from the sample. The final oxide is obtained at 410 °C (Fig. 8-2). The total mass loss observed during thermal decomposition of the sample is in good agreement with the calculated loss required to convert Bi2Ti3(sal)8(Hsal)2·3CH2Cl2 to the corresponding metal oxide (ca. 75%, obs. 76%).
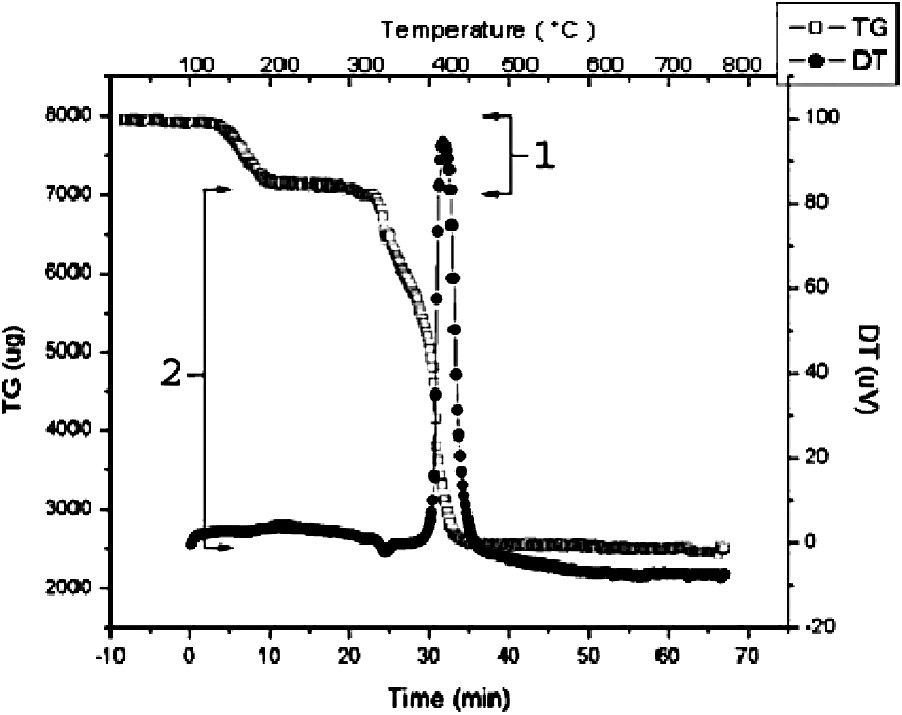
TGA/DTA traces of the thermal decomposition of Bi2Ti3(sal)8(Hsal)2 × 3 CH2Cl2 in air.
As expected, the thermal decomposition of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O was very similar to what was observed for Bi2Ti3(sal)8(Hsal)2. In this case, the thermogram shows a quick mass loss of 9.7% starting at approximately 35 °C, again correlating to the loss of lattice solvent. Decomposition of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O begins at approximately 270 °C with a mass loss of 42%. This is followed by a second mass loss at 390 °C of 19%. Conversion of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O completely to metal oxides, requires the 60% mass loss from the sample. The final two steps of the thermal decomposition of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O represent a 61% mass loss, which agrees with the expected value.
For both Bi2Ti4(sal)10(Hsal)(OiPr)·H2O and Bi2Ti3(sal)8(Hsal)2, it was observed that the materials produced at the end point of thermal decomposition were amorphous and more strenuous conditions were required to induce crystallization. Analysis of the complexes that have been annealed for 2 h at 750 °C reveals that they are composed of a mixture of oxide phases (Figs. 9 and 10). The oxide produced from Bi2Ti4(sal)10(Hsal)(OiPr)·H2O is principally composed of the bismuth-rich oxide Bi4Ti3O12, with the anticipated product Bi2Ti4O11 only detected in ca. 30% relative abundance as estimated from powder X-ray diffraction. Similarly, Bi2Ti3(sal)8(Hsal)2 decomposes on heating to produce Bi4Ti3O12 and Bi2Ti4O11 in a ca. 90:10 ratio also as estimated from powder X-ray diffraction. The composition and ratio of the phases observed on decomposition the molecular precursors Bi2Ti4(sal)10(Hsal)(OiPr)·H2O and Bi2Ti3(sal)8(Hsal)2 agree both with what is predicted by the published binary phase diagrams on this system, and by the composition of the individual molecular complexes themselves [34].

Powder X-ray diffraction spectrum of the oxide produced from the thermal decomposition of Bi2Ti4(sal)10(Hsal)(OiPr)·H2O for 2 h at 750 °C. The phases present are Bi4Ti3O12 (#) and Bi2Ti4O11 (*).
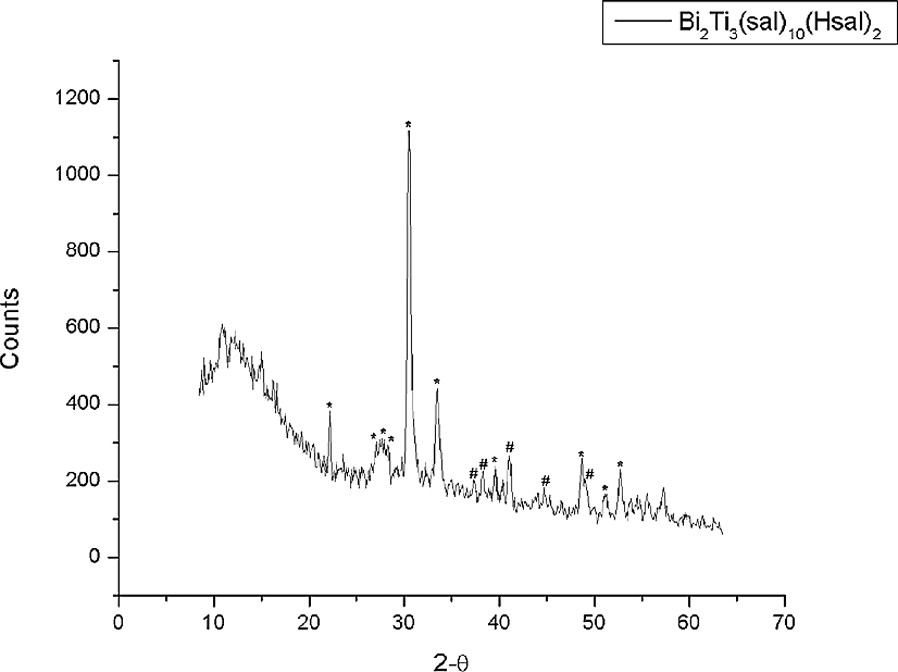
Powder X-ray diffraction spectrum of the oxide produced from the thermal decomposition of Bi2Ti3(sal)8(Hsal)2 for 2 h at 750 °C. The phases present are Bi4Ti3O12 (#) and Bi2Ti4O11 (*).
3.2 Thermal decomposition studies of niobium and tantalum alkoxide derived complexes
Recently it has been reported that the simple binary oxides BiNbO4 and BiTaO4 are ferroelectric when the thickness of the material is less than 250 nm and antiferroelectric materials when the oxides are thicker than this [35]. Additionally, it also been reported that BiNbO4 is a promising photocatalyst for the decomposition of water. As discussed in Section 2.2, we have developed several compounds whose stoichiometry make them promising precursors for the formation of these materials, and consequently we have made a more detailed investigation of the effects that the time and temperature of the sintering process have on the oxide produced from the thermal decomposition of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O. We have found that the complex decomposes on heating in air for 2 h at 750 °C to produce the metastable high temperature form of BiNbO4 (triclinic setting, BiNbO4-H) as the major crystalline phase (Figs. 11 and 12). The balance of the material was found to be the orthorhombic low temperature form of the bimetallic oxide, BiNbO4-L [36].

Powder X-ray diffraction pattern of the oxide of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O produced from various calcination temperatures for 2 h (A) and from calcination at 750 °C for various lengths of time (B). The phases present are BiNbO4–H (•), BiNbO4-L (♦) and Bi5Nb3O15 (*). (Used by permission of the American Chemical Society).
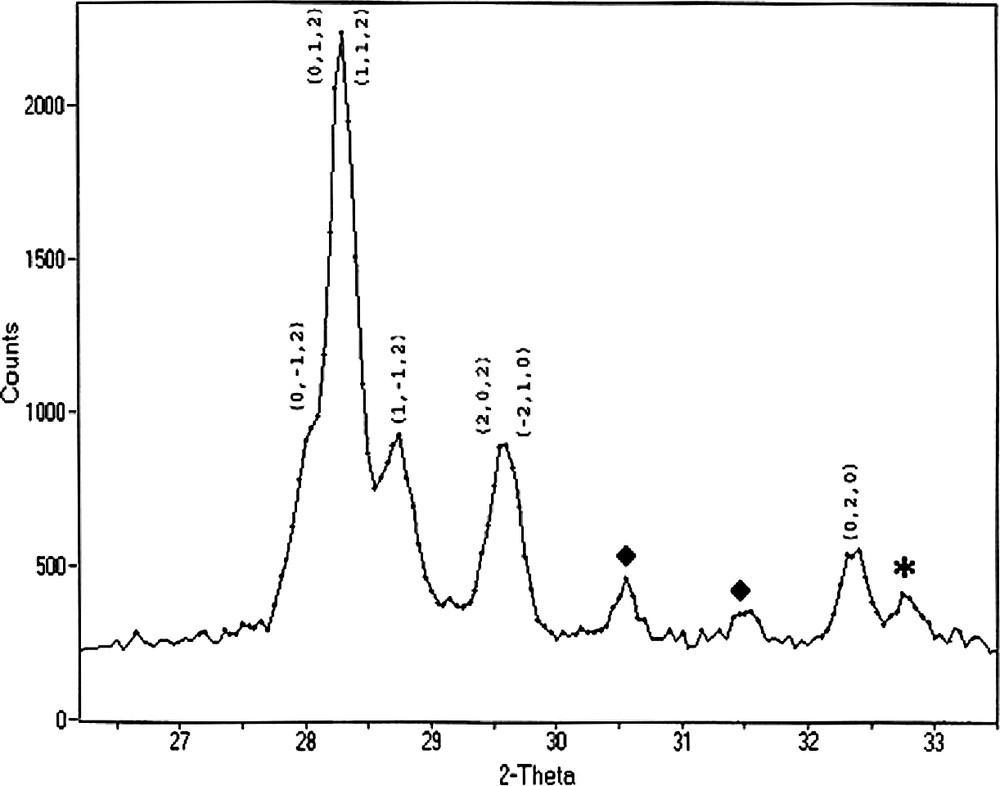
Indexed powder X-ray diffraction pattern of the oxide of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O produced after heating the molecular precursor for 2 h at 650 °C in air. Indexed peaks relate to the high temperature (triclinic) phase of BiNbO4. Peaks marked with an ‘♦’ relate to the low temperature (orthorhombic) phase of the same material, while peaks marked with an ‘*’ relate to Bi5Nb3O15. (Used by permission of the American Chemical Society).
Under the conditions detailed above, the high and low temperature phases in the material were found to be present in an approximately 4:1 ratio. Increasing the temperature of the sintering process favors conversion of BiNbO4-H into BiNbO4-L, along with concomitant disproportionation of the bimetallic oxide into Bi5Nb3O15 and Nb2O5. Similarly, increasing the sintering time of the sample was found to favor the conversion of BiNbO4-H into BiNbO4-L, although to a smaller extent. Upon either increasing the reaction temperature from 650 °C to 850 °C or the reaction time from 2 to 6 h, the (0 4 0) and the (0 0 2) reflections of the BiNbO4-L phase increase in intensity and sharpness, while the overlapping (2 0 2) and (2 1 0) reflections of the BiNbO4-H become more diffuse or disappear entirely. These results suggest that the initial formation of BiNbO4 is kinetically controlled. However, once the bimetallic oxide is formed, the thermodynamically more stable BiNbO4-L is favored and continued reaction and crystallization of the material will favor the formation of this product at the expense of the BiNbO4-H. This behavior has been observed in several other binary oxide systems that are produced using classical or modified sol–gel techniques [37].
As noted above, extended reaction of the BiNbO4-H results in the disproportionation to Bi5Nb3O15 and Nb2O5 as well as conversion to BiNbO4-L. It seems likely that the disproportionation products are formed from the BiNbO4-H. However, at this point it is not clear whether these products form directly from BiNbO4-H, or form via the intermediacy of BiNbO4-L or, possibly, by both pathways. It does appear, however, that the Bi5Nb3O15 and Nb2O5 phases do not result directly from conversion of the molecular precursor Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2. Other possible pathways for the formation of the disproportionation products involving either volatilization of bismuth oxide during decomposition of the molecular precursor or possible reaction of the oxide mixture with the ceramic crucible to form a sillenite-related phase of approximate composition Bi12Nb0.29O18.7–x are possible, but were not observed here [38]. We have found that sintering the samples at 600 °C effectively inhibits the production of disproportionation products and BiNbO4-L so that BiNbO4-H is produced as the only crystalline phase. These results agree with other findings on the formation of metastable states in bimetallic oxide systems [38,39].
Several techniques including BET, SEM and EDX have been used to study the morphology of the oxides produced from the thermal decomposition of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O and Bi2Ta2(μ-O)(sal)4(Hsal)4(OEt)2·H2O. Pyrolysis of the solid-state compounds produces a porous oxide, probably owing to the concomitant high energy of the reaction environment and oxidation of the organic ligands. SEM analysis of the materials produced in this manner reveal the presence of numerous pores and channels permeating the oxide (Fig. 13). The products of pyrolysis at low temperatures (i.e. < 750 °C) are found to be composed of aggregated particles indicating that the thermal decomposition process of the complexes likely proceeds through the formation of bimetallic oxide particles that then aggregate to produce the bulk material (Fig. 14). BET surface area analysis reveals that the surface areas of the oxides of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O and Bi2Ta2(μ-O)(sal)4(Hsal)4(OEt)2·H2O are 3.99 and 6.62 m2/g, respectively (instrumental standard deviation = 1.3%). These results indicate that the majority of the pores in the oxide are macroscopic, producing a material with both a low density and a relatively low surface area.

SEM image of the bulk pyrolysis product of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O produced after 2 h at 750 °C. (Used by permission of the American Chemical Society).

SEM image of a cross section of the bulk pyrolysis product of Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O produced after 2 h at 750 °C detailing the presence of oxide particles on the order of 150 nm in diameter.
The utility of the molecular precursors in facilitating the formation of the bimetallic oxides is clearly demonstrated in comparing the conditions required for the production of the high and low temperature phases by conventional methods to what was required in this work. For example, direct pyrolysis of the molecular complex Bi2Nb2(μ-O)(sal)4(Hsal)4(OEt)2·H2O allows for the formation of crystalline BiNbO4-H at temperatures of 650 °C in 2 h. In contrast, reported solid-state syntheses require sintering periods of 24–240 h at 1173 °C to achieve the same material [38].
3.3 Thermal decomposition studies of Lewis acid–base adducts of bismuth salicylate
Detailed investigations of the thermal decomposition of the complexes Bi2(Hsal)6·M(acac)3 (M = Al, Cr, Fe, V, Co) were pre-formed using both TGA/DTA and powder X-ray diffraction. In particular, we were interested in investigating the stability of the complexes during thermolysis against metal segregation, which could have deleterious effects on the chemical and physical properties of the resulting materials. In these systems, two likely routes to metal segregation can be envisioned to occur. In one case, the diketonate species could be liberated from the bismuth carboxylate – likely through a sublimation event, so that the resulting oxide is compositionally rich in the main group element compared to what is predicted from the molecular precursor. Alternately, it is possible that the different thermolytic stability of the diketonate and the carboxylate complexes would result in thermal decomposition of the main group element and transition element at different temperatures and consequent metal segregation.
Representative thermograms from the TGA/DTA studies of the complexes Bi2(Hsal)6·M(acac)3 (M = Al, V) are provided in Figs. 15 and 16. The general features of the TGA traces resemble what was observed in the pyrolysis of the alkoxide derived compounds, with thermal decomposition of the organic ligands beginning at approximately 180 °C. The decomposition process ends in most cases with an abrupt loss of mass, which occurs simultaneously with a strong exotherm in the DTA trace. The origin of this exotherm has been tentatively assigned to crystallization event in the resulting bimetallic oxide lattice. The DTA trace of the vanadium complex is remarkable because it contains two additional exothermic events. These peaks have been correlated to the step-wise oxidation of the transition metal center from V3+, which is present in the molecular precursor, through V4+ (Fig. 16-1) to the V5+ (Figs. 16-2) ion that is present in the bimetallic oxide Bi2VO11. In both cases, the observed mass loss of the samples agreed with quantitative conversion of the molecular precursors to the bimetallic oxides.
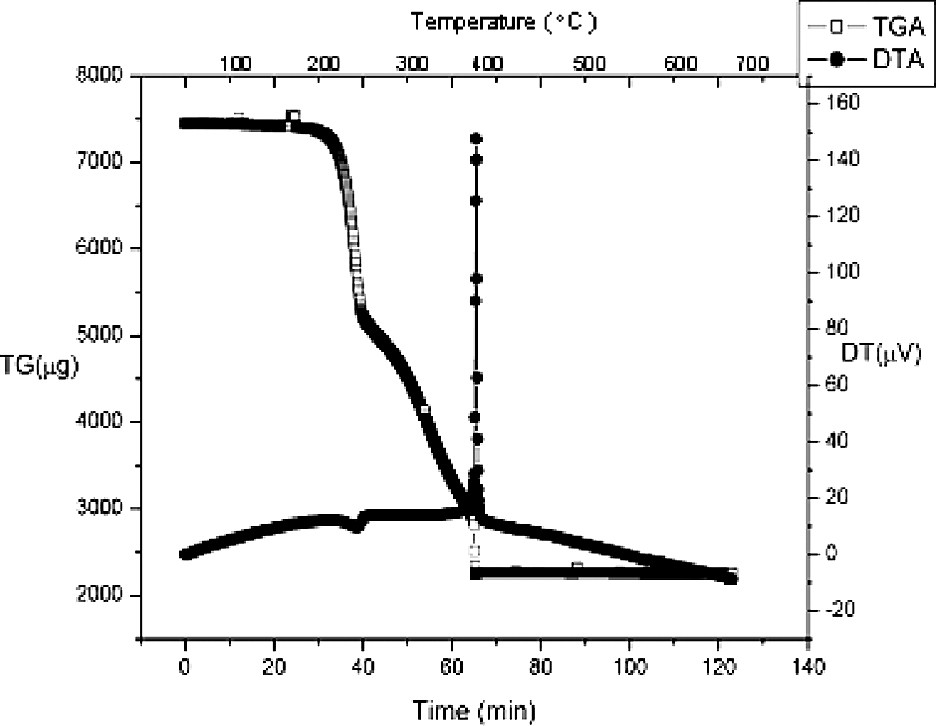
TGA/DTA traces of the thermal decomposition of Bi2(Hsal)6·Al(acac)3 in air.

TGA/DTA traces of the thermal decomposition of Bi2(Hsal)6·V(acac)3 in air.
Confirmation of the results from the DTA/TGA investigations has been obtained through interrogation of the resulting decomposition products by powder X-ray diffraction studies. In all cases, the phases present in the oxide correlate exactly to what is predicted by the binary phase diagrams for these oxide systems. In the case, of Bi2(Hsal)6·V(acac)3, the anticipated oxide Bi4V2O11 was observed to be the only crystalline material present (Fig. 17). In the case of Bi2(Hsal)6·Al(acac)3, thermal decomposition produced a 3:1 mixture of tetragonal Bi2O3 and Bi2Al4O9 (Fig. 18). It is important to note that in both cases, the crystalline binary oxides produced in this manner was obtained at conditions significantly milder than what has been previously employed for the synthesis of these materials (e.g 100 h at 1000 °C) [38]. These results clearly demonstrate the potential for these heterobimetallic coordination complexes to produce binary or ternary oxides under much more accessible conditions than those required for traditional solid-state syntheses.

Powder X-ray diffraction spectrum of Bi4V2O11 produced from the thermal decomposition of Bi2(Hsal)6·V(acac)3. Peaks marked with a * represent an unidentified trace impurity.

Powder X-ray diffraction pattern of the material produced from the thermal decomposition of Bi2(Hsal)6·Al(acac)3. Peaks marked with a ‘#’ represent the tetragonal phase of Bi2O3, while peaks marked with a ‘•’ represent Bi2Al4O9.
Similar approaches were employed to study the thermal decomposition of Bi(Hsal)3·Cu(salen), Bi(Hsal)3·Ni(salen) and Bi(Hsal)(sal)·V(O)(salen*). The bimetallic compound Bi(Hsal)(sal)·V(O)(salen*) begins to decompose at 200 °C and the final oxide is obtained at 430 °C. The thermal decomposition of the complexes was found to proceed under mild conditions, with the decomposition of the complex Bi(Hsal)(sal)·V(O)(salen*) complete at almost 70 °C less than what was observed for some of the bismuth–titanium complexes discussed earlier. As with the diketonate complex Bi2(Hsal)6·V(acac)3, the decomposition of Bi(Hsal)(sal)·V(O)(salen*) occurs in several steps, and simultaneous differential thermal analysis of the material revealed the presence of two strong exotherms at 368 °C and 410 °C. It seems likely that the exothermic event observed at 368 °C relates to a conversion of the V4+ species present in the molecular precursor to the V5+ ions that are present in the final oxide.
The product produced via the pyrolysis of Bi(Hsal)(sal)·V(O)(salen*) has been explored by powder X-ray diffraction and by SEM. Powder X-ray diffraction studies demonstrated that heating a sample of Bi(Hsal)(sal)·V(O)(salen*) for 2 h at 450 °C produces monoclinic BiVO4 as the only crystalline phase (Fig. 19). This stoichiometry of the oxide agrees with what is present in the molecular precursor and indicates that metal segregation does not occur during the pyrolysis event. Investigation of the oxides by SEM illustrates that the material produced in this manner is composed of highly aggregated oxide spheres (Fig. 20A). The isolation of discrete dispersable oxide spheres of BiVO4 by direct thermolysis of Bi(Hsal)(sal)·V(O)(salen*) has not been achieved. Increasing the annealing conditions of the material to 24 h at 750 °C did not result in any change in the phases present in the sample. However, the average diameter of the particles present in the sample was found to increase from 110 nm to approximately 65 μm (Fig. 20B). In this case, the morphology of the sample has come to resemble that which is commonly associated with the monoclinic phase of BiVO4.
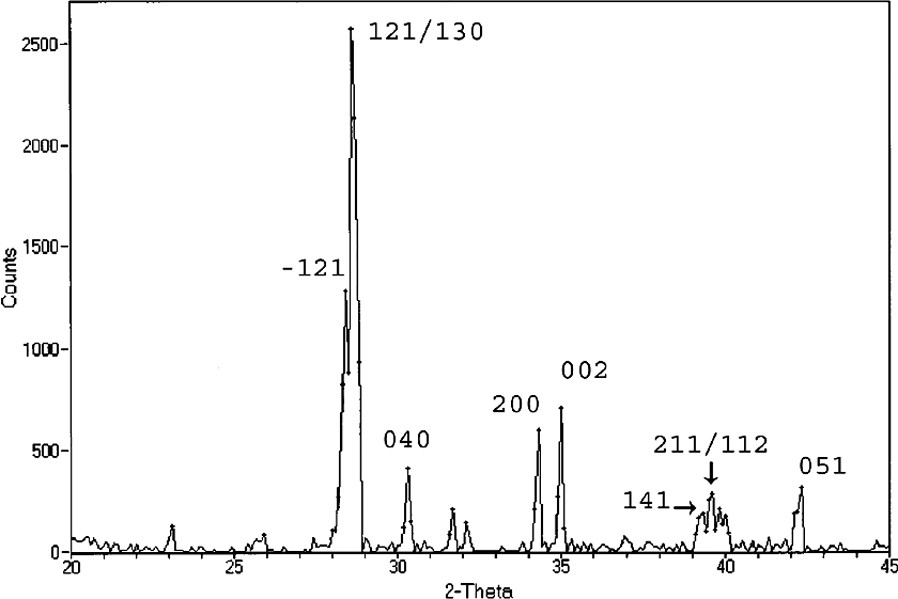
Indexed powder X-ray diffraction data of the material produced from the thermal decomposition of Bi(Hsal)(sal)·V(O)(salen*). All of the peaks are indexed to the monoclinic form of BiVO4. (Used by permission of the American Chemical Society).

SEM image of the pyrolysis product of Bi(Hsal)(sal)·V(O)(salen*) produced after heating the molecular precursor for 15 min at 450 °C (a) and for 2 h at 750 °C (b). The insert figure details a single bimetallic oxide sphere, with a diameter of approximately 110 nm, produced during the course of pyrolysis of Bi(Hsal)(sal)·V(O)(salen*). (Used by permission of the American Chemical Society).
Similar investigations have been performed on compounds Bi(Hsal)3·Cu(salen) and Bi(Hsal)3·Ni(salen), due not only to the fact that the doped samples of bismuth vanadate have been shown to be extremely efficient oxide ion conductors, but also to the fact that the binary oxides produced from these mixtures are potentially useful as well. For example, bismuth–nickel oxides are being explored as catalysts for the dehydrogenation of hydrocarbons [40] and for the transalkylation of aromatic hydrocarbon systems [41]. Similarly, bismuth–copper oxides have been investigated as catalysts for the synthesis of 1,4 butynediol from acetylene and formaldehyde [42] and for the removal of nitrogen oxides from exhaust streams [43]. However, the binary oxide systems of bismuth and the later transition metals have not been completely explored and some conflicting reports remain regarding the formation or stable phases of these compositions.
The thermal decomposition of the compounds Bi(Hsal)3·Cu(salen) and Bi(Hsal)3·Ni(salen), as monitored by TGA/DTA, is very similar to that of Bi(Hsal)(sal)·V(O)(salen*). The decomposition of the compounds begins at 190 °C and 170 °C, respectively, and is complete by 400 °C in both cases. Both compounds exhibit a large exothermic event in the differential thermal analysis that occurs immediately prior to the formation of the final decomposition product; similar to what is observed in Bi(Hsal)(sal)·V(O)(salen*) and, like Bi(Hsal)(sal)·V(O)(salen*), this has been assigned to a reorganization of the oxide lattice as essentially no mass is lost. SEM analysis reveals that Bi(Hsal)3·Ni(salen) decomposes to produce a large number of spheres with an average diameter of 275 nm (Fig. 21). The balance of the sample was observed to be amorphous material. Compound Bi(Hsal)3·Cu(salen) was found to behave similarly but produced significantly more amorphous material under identical decomposition conditions and the diameters of the oxide spheres that were produced were found to vary significantly more (175 nm–1 μm) and the particle morphology was less regular (Fig. 22). In both cases, the composition of the spheres has been probed by EDX and both metal species were found to be present in the sample.

SEM image of spheres produced from the thermal decomposition of Bi(Hsal)3∙Ni(salen) at 450 °C for 2 h. (Used by permission of the American Chemical Society).

SEM image of a ‘sphere’ produced from the thermal decomposition of Bi(Hsal)3∙Cu(salen) at 450 °C for 2 h.
Attempts to crystallize the materials produced through the thermal decomposition of Bi(Hsal)3·Cu(salen) and Bi(Hsal)3·Ni(salen) by more strenuous heating resulted in the formation of a melt. As a result of this unexpected behavior, samples of the initial decomposition product of Bi(Hsal)3·Ni(salen) was studied by TEM and selected area electron diffraction (SAED). Additionally, the materials produced during the course of thermal decomposition of the molecular precursor were analyzed by variable temperature powder X-ray diffraction. Transmission electron microscopy analysis showed that the spheres are primarily composed of NiBi3, but also contained a significant number of 7–10 nm oxide crystallites, which were found to have a face centered cubic (fcc) habit and were subsequently identified as NiO by electron diffraction (Fig. 23).
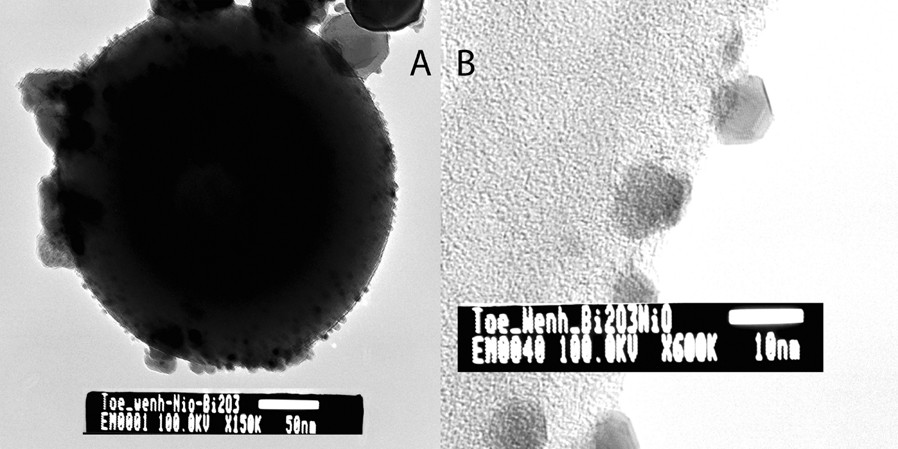
Transmission electron micrograph of a NiBi3 sphere (A) and of the NiO nanocrystals (B) produced during the thermal decomposition of Bi(Hsal)3∙Ni(salen) at 450 °C for 2 h.
Temperature programmed in-situ X-ray Diffraction of Bi(Hsal)3·Ni(salen) in the temperature range of ambient to 450 °C shows that the complex initially decomposes to produce an amorphous material, as suggested by the electron microscopy studies. However, on heating to 350 °C the orthorhombic CaLiSi2-type binary alloy NiBi3 was identified as the major crystalline component of the product mixture. Above 450 °C this phase melts (mp 469 °C) leaving an as-yet-unidentified material as the only solid crystalline phase in the sample. It is important to note, that the in-situ X-ray studies did not show any evidence of NiO, though it is likely that this is due to the crystallite size being below the threshold required for successful X-ray diffraction. Nevertheless it demonstrates clearly that the decomposition of the parent Bi(Hsal)3·Ni(salen) complex follows a different decomposition pathway compared to the other heterometallic complexes previously discussed, in this case possibly involving decarboxylation of the salicylate ligands. Such chemistry has been previously reported for the formation of main group organometallic species including the trinuclear complex Hg3(C6F4)3 [44]. It must be stressed that the decomposition of the heterobimetallic complex does not appear to generate amorphous oxides as previously suggested [45] but instead produces an anticorrosive NiBi3 alloy that resists further oxidation even upon annealing under air (Fig. 24).
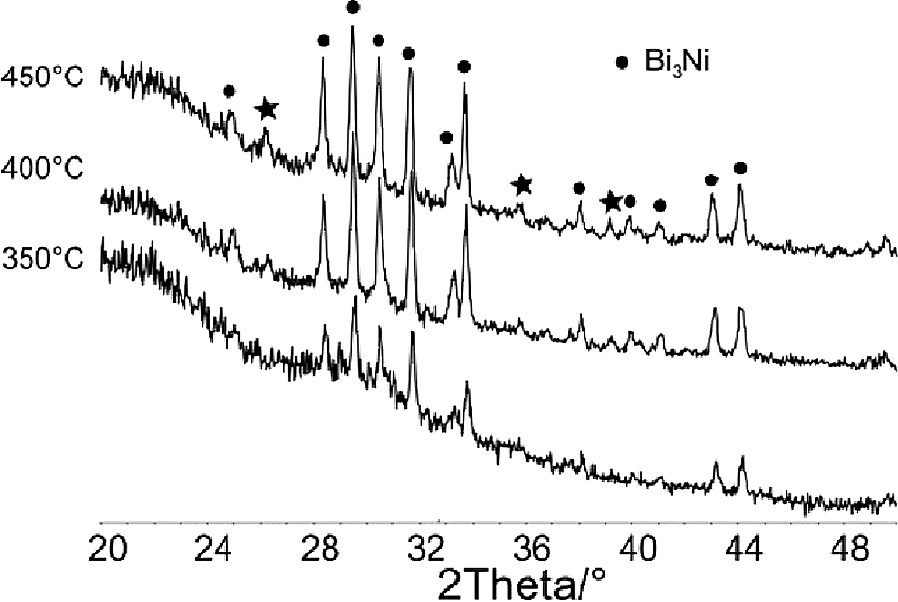
The patterns shown were collected at 350 °C, 400 °C, 450 °C. Between 350 and 450 °C, Bi3Ni appears to be the main crystalline phase. At 450 °C, the reflections of a new phase are observed, which cannot be assigned. These reflections are marked with a star. Above 450 °C Bi3Ni disappears whereas the reflections of the unknown phase remain.
We have investigated whether multi-metallic oxides containing bismuth, vanadium and either copper or nickel are accessible by direct doping of the molecular precursor Bi(Hsal)(sal)·V(O)(salen*) with samples of Bi(Hsal)3·Cu(salen) or Bi(Hsal)3·Ni(salen) in various ratios prior to thermal decomposition of the samples. A highly crystalline purple material was obtained from the pyrolysis of Bi(Hsal)3·Cu(salen) in the presence of Bi(Hsal)(sal)·V(O)(salen*) for 2 h at 750 °C. A similar experiment utilizing Bi(Hsal)3·Ni(salen) produced a reddish oxide product. SEM analysis of the products of decomposition reveals that both samples possess well-formed crystallites that are not observed in the thermal decomposition of the molecular precursors alone. The ratio of complexes employed in these experiments was initially intended to approximate the transition metal ratio of the Bi2Cu0.1V0.9O5.5–δ systems, although the product should be lean in bismuth due to the stoichiometry of the molecular precursors. In all cases, the product oxide acquired from heating the mixtures at 750 °C for 2 h was found to be composed of a mixture of the anticipated BiVO4 and a transition metal doped Bi2VO5.5-type phase.
Determination of the fate of all of the metal species in these experiments is difficult as the powder X-ray diffraction patterns of the termetallic BiMVOx materials are nearly identical to that of Bi2VO5.5 [45]. In order to more effectively investigate the composition of the oxides produced in this manner, the ratio of the molecular compounds was changed from 10:1 to 1:1. Pyrolysis of this mixture under identical conditions to the initial experiments resulted in the formation of a Bi2VO5.5 type material as the only crystalline bismuth-containing phase. It is important to note that where we now have evidence that the thermal decomposition of the bismuth–nickel complex by itself results in the formation of alloy compounds—presumably through a decarboxylation reaction of the salicylate ligand. Inclusion of vanadium in the mixture effectively quenches this process and the ternary oxides are formed in high yields. The bimetallic compounds presented here can be used to synthesize the termetallic system Bi2VxM1–xO5.5–δ (M = Cu, Ni) by simple mixing of the molecular precursors prior to pyrolysis. In all cases, the conditions required for conversion of the molecular precursors to the oxide materials are significantly less than what is required for similar solid-state syntheses.
3.4 Hydrolytic decomposition studies
Smaller bimetallic nanoparticles have been synthesized by hydrolysis of the molecular precursors in the presence of the stabilizing agents stearic acid and diphenyl ether. The particles that are isolated from this reaction have an average diameter of approximately 40 nm (Fig. 25). The materials produced in this manner do not possess the regular spherical morphology typifying the thermal decomposition products, but these materials were found to be repeatedly dispersible in a variety of solvents, although some aggregation of the particles is observed on isolation of the materials. This aggregation has been attributed to loss of the stabilizing diphenyl ether from the surface of the oxide particle. Analysis of the oxides produced using hydrolytic approaches by EDX shows that, similarly to the thermal decomposition methods, the metal stoichiometry of the molecular precursor is conserved in its transformation to the oxide material [45].

SEM image of the oxide particles produced from the hydrolytic decomposition of Bi(Hsal)(sal)·V(O)(salen*) in the presence of stearic acid. The dimensions of the particles are in the order of 30–50 nm. The inset image details a cluster of oxide particles produced in this manner. (Used by permission of the American Chemical Society).
4 Conclusions and future directions
In this work we have illustrated that both bifunctional ligands and Lewis acid–base adduct formation represent viable synthetic routes to the formation of heterobimetallic coordination complexes of bismuth in excellent yields. The complexes formed by these approaches smoothly decompose under thermolysis to generate the anticipated bimetallic oxides under very mild conditions – particularly when compared to traditional solid-state approaches. When late transition metals such as copper and nickel are included in the heterobimetallic coordination complexes, thermolysis unexpectedly favors the formation of binary alloys as opposed oxide based materials. These oxides are readily redispersible in polar solvents.
The residual functionality present in these complexes presents the possibility of further functionalization and derivatization. The step-wise manner in which many of these complexes can be constructed – particularly through Lewis acid–base chemistry – offers the tantalizing possibility that still more chemically complex species, including ternary compounds can be accessed through rational, designed wet-chemical approaches.
Acknowledgments
Authors would like to thank the NSF for grant CHE-9983352 and the Robert Welch Fondation for their financial support and Dr. Claudia Weidenthaler from the MPI for Kohlenforschung for fruitful collaboration in the analyses of the these materials.