

1 Introduction
The study of relatively simple organic molecules under non-ambient conditions is a vital source of information regarding the underlying physical forces that hold these molecules together. By varying the thermodynamic conditions under which the crystal structure is studied, vital information can be gained which promises to increase our understanding of the forces holding such molecules together. Such structural information also provides vital benchmarks for the parameterisation of theoretical and computational methods for the study of molecular structure, and also to the interpretation of spectroscopic studies.
We focus in this brief account on the application of neutron diffraction to the longstanding but still vital problem of the geometry, proton position and shape of the potential well in short hydrogen bonds in organic crystal structures. An understanding of such hydrogen bonds is a longstanding problem in crystal chemistry [1]. They have been widely studied and have been implicated, for example, as the structural feature responsible for various types of critical phenomena in the solid state, including ferroelectric phase transitions [2].
In general the formation of a D–H···A hydrogen bond is expected to change the potential energy surface for the D−H group from a single minimum to a symmetrical or unsymmetrical double minimum. This simple picture, however, does not hold for strong hydrogen bonds, which typically have O···O distances in the range 2.4–2.55 Å. Strong hydrogen bonds have been subjected to much analysis by single crystal neutron diffraction, as they are occasionally found to be close to symmetric, and often have elongated D−H covalent bonds and consequently shorter H···A separations. The positions of the hydrogen atoms are, however, also strongly affected by the crystalline environment. Neutron single crystal diffraction provides an ideal tool for examining proton positions and proton behaviour in such cases. In particular, the technique allows refinement of the anisotropic displacement parameters of hydrogen atoms, yielding information on the interatomic potentials experienced by these atoms. The most accurate early neutron diffraction studies, whose conclusions remain valid today, led to the view that the potential energy curve in fact becomes flattened close to its minimum and the proton vibrates anharmonically in this curve. This view is supported by the fact that protons in short hydrogen bonds frequently exhibit atomic displacement parameters (ADPs) that are elongated in a direction along the hydrogen bond. The potentials in which the proton sits can also be strongly affected by local crystal environment, so that these are usually not only anharmonic but also asymmetric.
The analysis and understanding of such hydrogen bonds is hampered by these subtle effects in the local shape of the potential, manifest in diffraction experiments in the shapes of the thermal ellipsoids. In refinements, it is similarly found to be difficult to distinguish various structural models. For example, even high quality diffraction data may not allow a distinction to be made between the case where the proton vibrates about a single potential minimum and that where it randomly occupies alternative sites slightly on either side of the mid-point [3]. This difficulty is exacerbated in short ‘centred’ O−H−O hydrogen bonds where any split proton sites would be only around 0.2 Å apart, to be contrasted with the classic situation of a disordered proton, for example in normal ice Ih or in benzoic acid [4,5], where the disordered proton positions are separated by some 0.6–0.7 Å. There have thus been arguments made that partial proton disorder in such systems is possible at O···O separations of 2.50 Å (between sites corresponding to unequal O–H and H···O distances) while symmetrically positioned protons must necessarily become the norm as this distance approaches 2.40 Å.
The aim of much of our recent neutron diffraction work in this area has been to use variable temperature studies to help characterise the behaviour of protons in such situations – by increasing the temperature, the proton has more energy and so in principle is more free to explore the shape of the potential well. In this view, the position and thermal parameters of the proton at each temperature can give a fairly subtle indication of how the potential is acting to determine the behaviour of the proton. In previous work, by measuring a structure at a range of temperatures, we have been revealing important extra information regarding the behaviour of these hydrogen atoms and the potentials in which they sit. We have developed and applied rapid data collection methods to the study of hydrogen atoms in a range of interesting chemical and crystal environments including those in librating groups [6], in short, strong hydrogen bonds [7,8] and in carboxylic acid dimers [5,9].
We report here briefly on some of these and related findings, and also on the extension of this work to the study of molecular materials with short hydrogen bonds (both intra- and intermolecular) under modest pressures (up to 5 kbar). Using neutron single crystal diffraction as a probe, and exploiting its ability to determine accurate hydrogen atom parameters, we aim to change the shape of the hydrogen bonding potential by applying high pressure. This is complementary to the application of variable temperature (in the absence of phase transitions), where the aim is to provide extra energy for the proton, allowing it more freedom to explore the shape of the potential well, and is thus a natural extension of previous variable temperature work [5–10]. However, we also note that in a solid-state (lattice) environment, the shape of the hydrogen bond potential can also evolve with temperature (see Section 4.2 below).
2 Neutron single-crystal diffraction under non-ambient conditions
2.1 The benefits of neutron diffraction in the study of molecular materials
Neutrons are a powerful probe of condensed matter and often the information available from neutron scattering is unavailable from other methods. The fact that neutrons are scattered by the nucleus, as opposed to X-rays, which scatter from the electrons, means that the scattering power does not vary monotonically with Z. This has several beneficial consequences for the application of neutron diffraction in the study of a wide range of materials. Elements with similar Z-values generally have substantially different scattering cross sections, offering a direct method of distinguishing neighbouring elements. Isotopes of the same element generally have substantially different scattering lengths for neutrons, making the technique of isotopic substitution very powerful – the most widely used isotopic substitution is that of deuterium for hydrogen; the very large difference between the scattering lengths of 2H (6.67 fm) and 1H (−3.74 fm) allows the use of contrast variation, allowing the scattering density of different parts of a molecule to be altered and highlighted. In addition, and of particular relevance here, it is possible to determine the parameters of light atoms, such as hydrogen, in the presence of heavier ones more accurately. For example, hydrogen atoms are determined around an order of magnitude more accurately with neutrons than X-rays in the presence of carbon atoms. The lack of a fall-off in scattering power as a function of scattering angle also gives neutron diffraction the ability to study structures to very high resolution, particularly when working at cryogenic temperatures.
Neutron single crystal diffraction [11] is the method of choice for defining precise and accurate parameters for hydrogen atoms, assuming sufficiently large (usually mm-sized) single crystals are available. It is thus the ideal technique for studies such as those outlined here.
2.2 New opportunities: neutron single crystal diffraction with large detector arrays
There has been a dramatic advance in the capabilities of neutron single crystal diffractometers over the past few years, making these valuable experiments available to a wider range of applications. The improvements in instrumentation have allowed neutron single crystal diffraction, intrinsically a flux-limited technique, to move towards allowing structure determination from small molecule samples such as those discussed here in just a few hours per data set. This allows the multi-T, multi-p approach to be adopted in these cases.
The most dramatic improvements in count rate in single crystal neutron diffraction have focused on large area detector Laue or quasi-Laue diffractometers; the detector solid angle coverage on these new instruments is 2π Sr or more, allowing for far more efficient measurement of diffraction patterns. These instruments include LADI and VIVALDI at the ILL reactor source in Grenoble [12], the former optimised for macromolecular structure determination and the latter for chemical crystallography. The same approach is adopted on the time-of-flight Laue diffractometer SXD (Fig. 1) at the ISIS spallation neutron source in the UK [13], on which the data discussed here were collected.
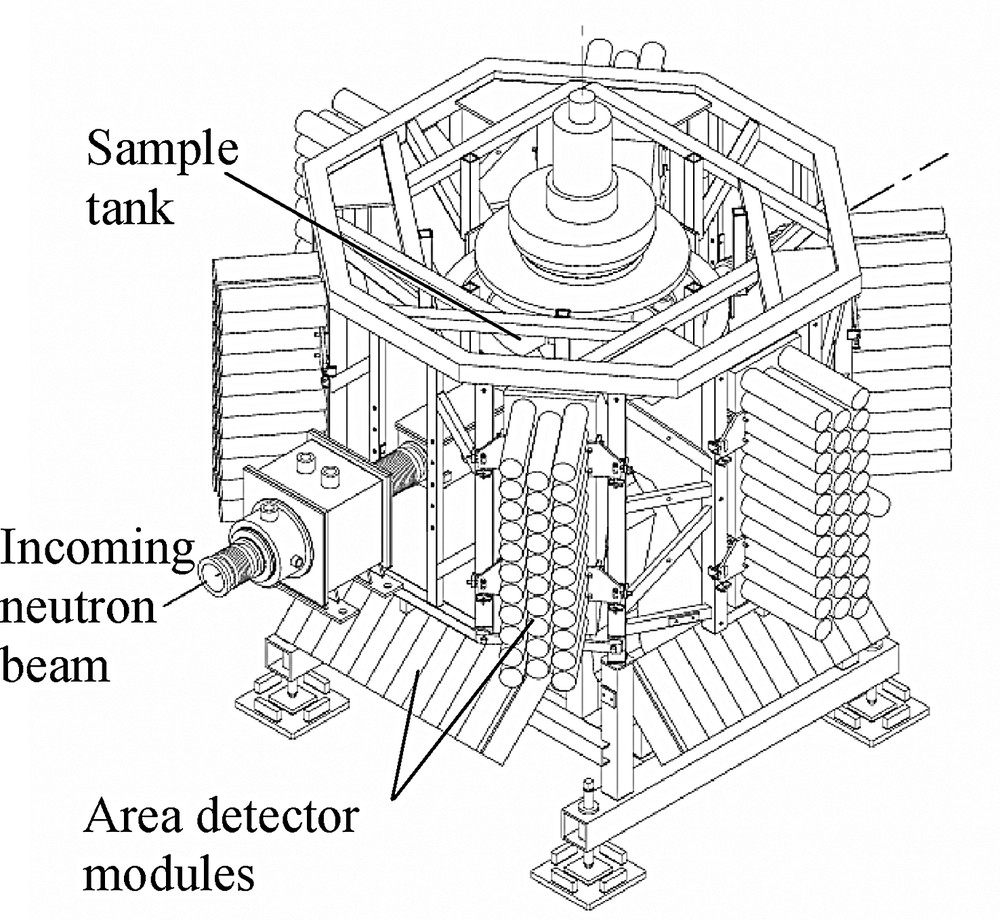
Schematic layout of SXD, the single crystal diffractometer at ISIS. Six detector modules are in the equatorial plane, four modules are tilted at 45 degrees below this plane and one is situated on the bottom of the instrument, directly underneath the sample position. The detector area coverage is ~2π Sr, > 50% of the available solid angle.
2.3 The high-pressure He-gas cell on SXD
The high-pressure cell used in this work is constructed of TiZr ‘null’-scattering alloy (the proportions of the two elements are chosen so as to give zero overall coherent (diffraction) scattering length. The design is based on that of a similar cell on the SCD instrument at IPNS, Argonne, but the cell has been redesigned to allow pressures of up to 5 kbar to be applied using He gas provided by an off-line intensifier. Pressure control is conveniently available to the user through a local ‘steering-wheel’ arrangement and the cell is now incorporated into the user programme on the instrument [14]. The internal diameter of the cell is ~1.7 mm, naturally restricting the size of crystal which can be used in the experiment. The design of the cell is of very open geometry, allowing the full extent of the 2π detector solid angle to be exploited in experiments. The cell can also be mounted on a closed-cycle refrigerator cooling device, allowing temperatures of as low as 30 K to be achieved simultaneously with the application of pressure. From a practical point of view, the mounting of the cell is restricted to a vertical geometry, so that in order to obtain representative three-dimensional single crystal diffraction data, a multiple-crystal sample (see below) is used as standard in this device.
2.4 Multiple-crystal data collection techniques
Given the geometrical constraints of the high p/low T set up used in these experiments, it is not always possible to collect a representative three-dimensional set of diffraction data from a single crystal mounting. In order to remove the need to remount crystals, a multiple-crystal data collection method was used in the experiments described here.
This method [15], now established as standard on the SXD instrument, involves the use of multiple single crystal samples to collect neutron diffraction data from low-symmetry molecular systems. There are several cases in which such an approach could be useful, including making data collection more rapid, in using several very small crystals to make data collection feasible or, as applied here, if the samples are quasi-randomly aligned in the beam, to obtain full three-dimensional diffraction data using very limited sample re-orientations. As applied here, this allows rotation about a single axis to yield sufficient data for good structural refinement.
The possibilities of collecting such multiple-crystal data sets are enhanced by the use of the time-of-flight Laue data collection method, as employed on SXD. The method allows for fully resolved three-dimensional data volumes to be accessed in a single measurement (a frame) from a stationary crystal-detector arrangement. The three-dimensional nature of the data thus gives the best possible chance of avoiding accidental overlap from reflections from the different crystals in the sample, since these can be separated in both space and time-of-flight (≡wavelength) on the detector.
3 Studies of molecular systems under varying (p,T) conditions
3.1 Urea–phosphoric acid
Urea has been widely used as a complexing agent for organic acids in investigations of hydrogen bonding phenomena. It is well known that the acidic H-atom in these complexes shows a varying degree of ‘transfer’ from one compound (the acid) to the other (urea). For example, in the urea–nitric acid (1:1) complex, an H-atom is transferred from nitric acid to the carbonyl oxygen of urea (Oacid···H = 1.591(3) Å, H–Ourea = 1.006(3) Å) to produce a salt, uronium nitrate [16]. This contrasts with the urea–oxalic acid (2:1) complex, in which the H-atom remains covalently bonded to oxalic acid (Oacid–H = 1.074(4) Å) and is hydrogen-bonded to urea (H···Ourea = 1.416(4) Å) [17]. Neutron diffraction analyses of urea–phosphoric acid (UPA) have shown a situation intermediate between these two extremes [18]. At room temperature, a near-symmetrical O···H···O arrangement is observed (Oacid···H = 1.223(6) Å, H···Ourea = 1.207(6) Å; (18)). Other data [19,20] suggest a greater degree of asymmetry in the arrangement at low temperatures, and we have shown in recent experiments that the proton in this hydrogen bond migrates as a function of temperature to a symmetric position [7].
Our previous extensive work on urea–phosphoric acid [7,20] has now been extended to high-pressure studies, to gain further knowledge of the behaviour of the ‘migrating’ proton, and the possible effect of the application of pressure on the centring of this atom. This centring is observed to occur with increasing temperature at ambient pressure in this symmetry-unrestricted strong hydrogen-bonded system. The experiment was carried out on SXD using four single crystals of average volume ~1 mm3 inside the pressure cell. Data were collected at four temperatures (295, 250, 200, 150 K) at each of two pressures (1 kbar and 2 kbar). Since the proton centring is evident at around room temperature at ambient pressure, it is of immediate interest to look at the 295-K, 2-kbar data to see if there is any induced effect on the proton position. Evidence is found for an asymmetrically located proton (difference in the two O–H distances). Following this trend with temperature (Fig. 2) shows this pattern to be repeated, with the application of 2-kbar pressure consistently increasing the asymmetry of the hydrogen atom position by about the same amount. This would suggest that a pressure of 2 kbar may indeed be significantly changing the shape of the potential, either directly or by influencing the external lattice effects.

The effect of pressure on the hydrogen bond asymmetry in urea–phosphoric acid. The difference in H···O and O–H distances is shown as a function of temperature for ambient pressure (●) and in our preliminary refinements of data collected at 2 kbar (□). Error bars show ± 1σ.
3.2 Potassium hydrogen maleate
The hydrogenmaleate ion [21] contains a short strong hydrogen bond where the position of the hydrogen atom is sensitive to the crystallographic environment. This group has been widely studied with varying cations, with observations of both symmetric [22,23] and asymmetric [24] hydrogen atom positions within the intramolecular hydrogen bond. The centred hydrogen bond in the imidazolium compound [22,23] is not constrained by crystallographic symmetry, while in the title compound the centred hydrogen atom is symmetry constrained [25,26], with the hydrogen atom sitting on a mirror plane. Neutron diffraction studies have confirmed the centring of this proton at a range of temperatures [27,28]. The experiments carried out here were aimed at investigating the possible effect of pressure on this symmetric hydrogen bond, and also in applying computational methods to the description of this system (see below).
Neutron diffraction data were collected on SXD under 8 sets of thermodynamic conditions in the range 30–295 K and 0–4 kbar (Fig. 3), with data collection times as low as 5 h per (T, p) point [29]. These measurements, yielding good coverage of p–T space, have allowed fully anisotropic refinements to be carried out on all data sets. The results of these refinements are shown in Fig. 3; these show the intramolecular hydrogen bonding in the structure to be essentially invariant in the range of conditions studied. The position of the hydrogen atom on the mirror plane is unaltered by the variation of temperature or pressure, there is no anomalous behaviour of the displacement ellipsoids and the molecule remains planar with no anomalous out-of-plane motions. The possibility of disorder of the proton about the mirror plane was also investigated in a series of difference Fourier maps; these again showed no evidence for asymmetry of the proton in the hydrogen bond.
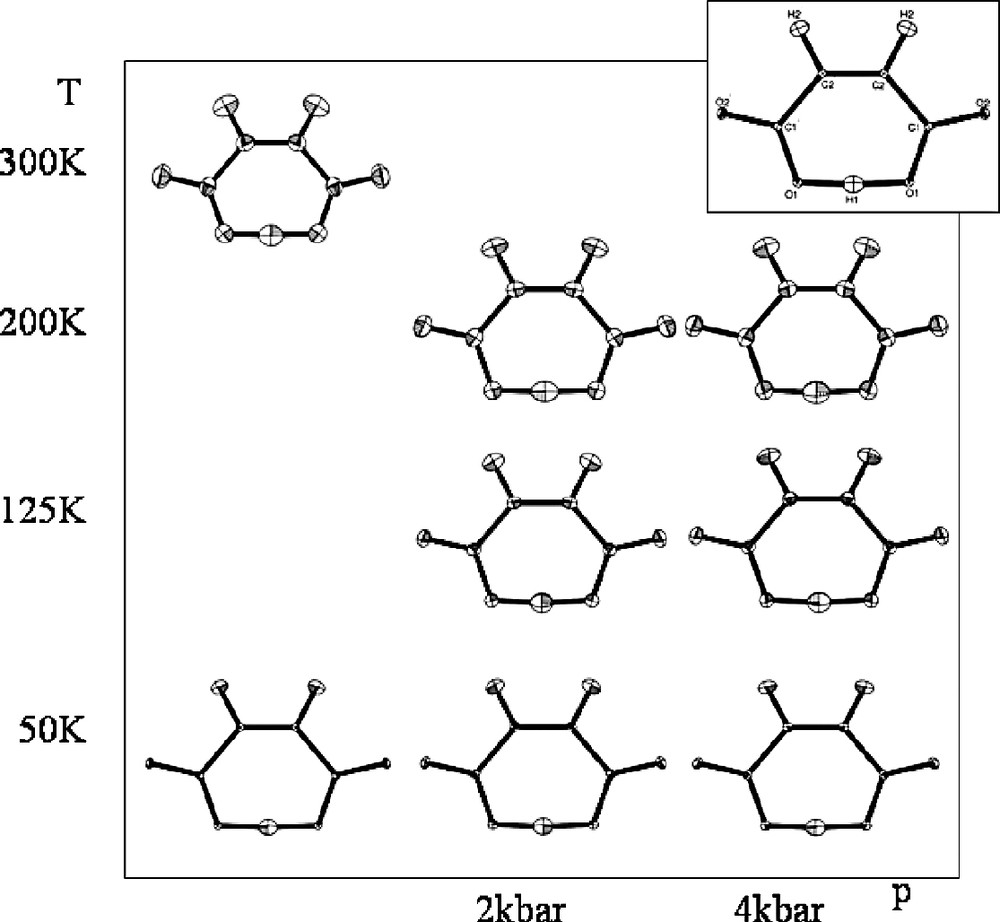
‘Structural-phase diagram’ showing the refined structure of the hydrogenmaleate anion in potassium hydrogen maleate, from single-crystal neutron diffraction data at a range of temperatures and pressures. Structures-drawn using ORTEP [30].
Although we have observed no change in the hydrogen atom position as a function of pressure, this work has shown that it is possible to carry out systematic investigations into the effects of variable temperature and variable pressure on molecular systems using single crystal neutron diffraction. Structural refinements and difference Fourier calculations give strong support to the model of a symmetric hydrogen atom, with no indication of asymmetry or disorder. These findings are borne out by periodic plane-wave calculations (see below).
3.3 Dibenzoylmethane
Dibenzoylmethane contains a short O···O intramolecular hydrogen bond. This compound was initially studied using X-ray diffraction where there was some contradiction about whether the hydrogen atom is asymmetric [31] or centred [32]. However, a neutron diffraction study [33] confirmed that dibenzoylmethane has a strong asymmetric intramolecular hydrogen bond (O···O distance: 2.459(4) Å, asymmetry = 0.199 Å) implying that the molecule must exist in the enol tautomer. The asymmetry results from the environment in the vicinity of the hydrogen bond, where there is a series of close contacts, just outside the van der Waals limit, undertaken by the acceptor oxygen atom. In order to investigate the temperature and pressure dependence of this asymmetric hydrogen bond, we undertook two separate experiments one probing the temperature evolution of the structure in detail from 100 to 305 K and the other probing the (T, p) evolution of the structure in the pressure cell (data collected at temperatures of 30 and 200 K for each of the pressures 0.4, 2, and 4 kbar). Typical crystal volumes were ~5 mm3 for the variable temperature study (single crystal) and ~1 mm3 multiple-crystal samples for the (T,p) measurements.
The results of the refinements are summarised in Fig. 4. The familiar features of a short, asymmetric hydrogen bond are present—a significant elongation of the O–H covalent bond and some elongation of the H atom thermal ellipsoid in the direction along the hydrogen bond. There seems to be little effect on the hydrogen bond geometry of changing either temperature or pressure. The thermal ellipsoid of the hydrogen atom may become less elongated as the pressure is increased, however, the data are not of sufficient precision for this to be conclusive. As with potassium hydrogen maleate, it would appear that this short intramolecular hydrogen bond is relatively robust to the application of both temperature and pressure changes in the range applied here.
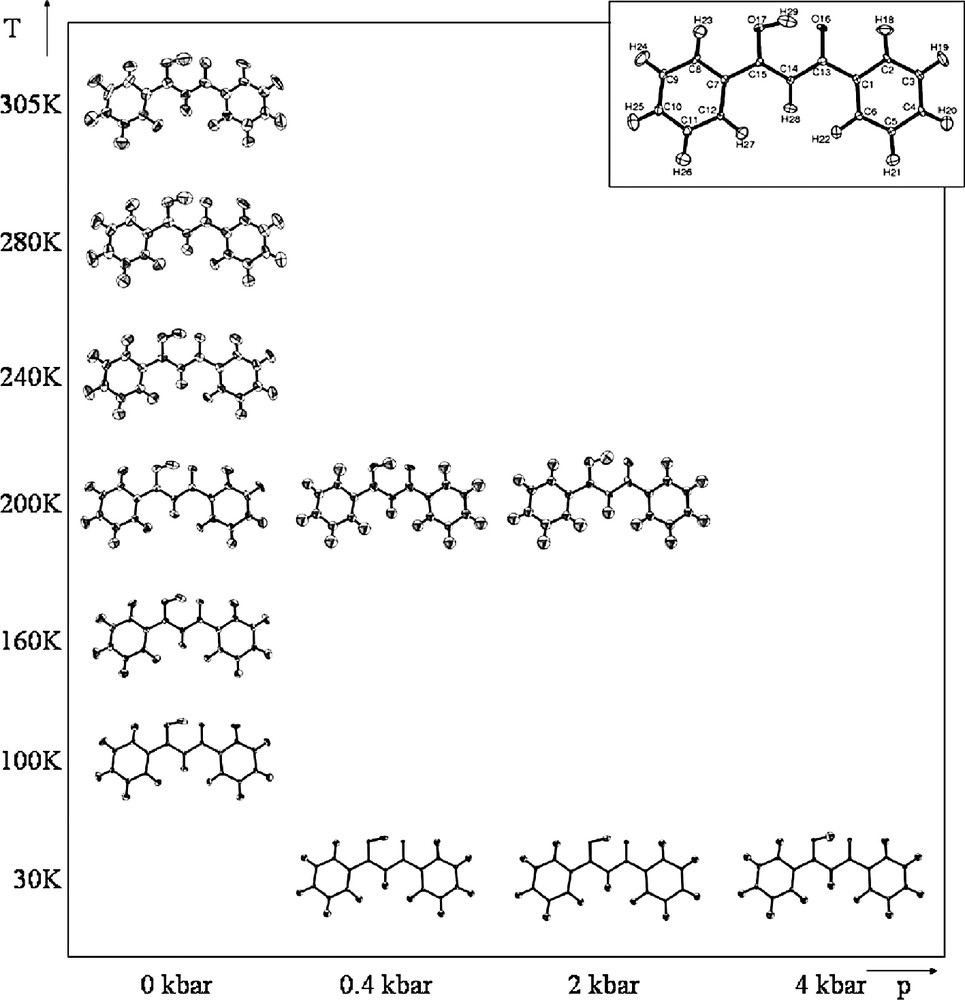
‘Structural-phase diagram’ showing the refined structure of dibenzoylmethane, from neutron diffraction data at a range of temperatures and pressures. Structures-drawn using ORTEP [30].
4 Complementary X-ray and computational studies
4.1 X-ray diffraction
The main thrust of our work in this area has for some time been the use of multi-temperature/multi-pressure single crystal neutron diffraction on appropriately large crystals of the compounds of interest, since neutron diffraction is the definitive structural technique for the description of hydrogen atoms. However, we have also re-examined the use of X-ray diffraction in this respect, and in particular the use of X-ray difference Fourier maps in tracking the evolution of hydrogen atom behaviour as external parameters are changed, an approach also extensively used by others with some considerable success [34]. Our initial expectations in this area were that revisiting this possibility would allow X-ray methods to be used both to screen out systems in which there appear to be no anomalous features worthy of study by neutron diffraction, and to indicate the temperature range of importance to any further investigations.
An example of the former is given by the study of potassium hydrogen phthalate [35], which contains a motif of longstanding interest, to us, the carboxylate group, and competition between possible hydrogen bonding motifs (with one COOH and one COO– group). In this case we collected both X-ray and neutron diffraction data at a range of temperatures. On examining the Fourier maps, it appears that the moderate/strong hydrogen bond within this structure does not display any anomalous behaviour, with neither proton migration nor any disorder observed at high temperatures in either the X-ray or the benchmark neutron data. The important consequence of this is that the results indicate that it may indeed be possible to use X-ray diffraction as a means of reliably screening such systems for anomalous hydrogen atom behaviour. If interesting behaviour such as proton migration or disorder is in any way indicated, neutron experiments could then be undertaken to study the complexes further. However, if no anomalous behaviour is observed then there is no need to embark on the more difficult neutron experiments.
The second aspect of using X-ray diffraction in this project is well illustrated by a variable temperature X-ray diffraction study of the structure of 2,4,6-trimethylbenzoic acid [36]. After initial attempts to acquire neutron diffraction data from this material were unsuccessful, X-ray experiments were employed to investigate the possibility of using the difference in C=O and C–O bond distances in the carboxylic acid dimeric motif as a fingerprint of possible hydrogen atom disorder. However, the results were more dramatically effective than initially expected, with difference Fourier sections through the (COOH)2 dimeric link showing a dominant peak corresponding to a single hydrogen atom position at 100 K, but a much more equal split site at 290 K. Measurements at intermediate temperatures (170 and 240 K) bear out this finding, and visually show an increasing trend in the occupation of the second site. We, therefore, have the clearest indication from our VT X-ray studies, of a proton disorder system in which the proton gradually orders onto one preferred site as the temperature is lowered. Indeed, in this case modelling the relative occupancies of the two possible proton sites based on the Fourier images allows us to use the simple Van't Hoff model to determine the energy difference between the two possible configurations in the crystal structure. This model yields an energy difference between the two configurations in the crystal structure of ΔH = 2.5(3) kJ.mol−1. This can be compared with the values of 0.50(4) kJ.mol−1 for benzoic acid and 1.64(9) kJ mol−1 for p-chlorobenzoic acid obtained previously from neutron diffraction data, and represents an unexpectedly quantitative result from X-ray diffraction of this system. While not a ‘short’ hydrogen bonding system, this is a fine illustration of the use of multi-temperature diffraction in describing structural evolution in a hydrogen-bonded system.
One final example of this approach is the complementary use of neutron and X-ray diffraction in describing short hydrogen-bonded systems. In this context we carried out variable temperature X-ray studies in the range 150–350 K on our model material urea–phosphoric acid with the aims of verifying that the migration observed in neutron diffraction can also be reliably observed using X-rays and to extract any additional information from the complementary X-ray and neutron measurements. The study was conspicuously successful in both of these aims [37]. Difference Fourier maps were calculated for each of the X-ray and neutron data sets at each temperature, and a comparison of the X-ray and neutron images showed that the migration of the proton with temperature is observed using both techniques; at 150 K the position of the hydrogen in these Fourier map images is significantly closer to the urea moiety, while at 350 K it is located at the centre of the hydrogen bond and is formally bound to neither moiety. However, a large difference is also observed in the position of the maximum density in the neutron and X-ray Fourier maps at low temperature. In the X-ray case the asymmetry is far more pronounced, reflecting the fact that in this case the electron density is being plotted and is influenced by the bonding present. Interestingly, the hydrogen atom is almost exactly centred at 300 K from the neutron data, whereas the electron density is not, suggesting that the hydrogen bonding density does not become centred until it is above room temperature. The consequence of this is that there is much larger apparent shift in the positions of the density maximum in the X-ray data as the temperature is increased compared to that observed in the neutron structure (~0.1 Å cf. ~0.04 Å); because we see the electron density in the former, the changing nature of the hydrogen bond is actually emphasised more in the X-ray than the neutron images.
4.2 Computational studies
In order to move towards our goal of understanding rather than simply measuring short hydrogen bonds in molecular materials, we have begun to employ plane-wave DFT calculations in trying to understand our solid-state structures.
In our first study of these systems we carried out pioneering modelling studies of the UPA hydrogen bond in the solid state [20]. Initially, a graded series of ab initio molecular orbital calculations were performed on an isolated dimer unit of UPA, with the starting geometry obtained from the low temperature neutron diffraction structure determination. These isolated molecule calculations do not reproduce the geometric features of interest in this complex and in particular, the highest level calculation produces an O···O separation of 2.61 Å (indicative of a normal length/strength hydrogen bond), along with O–H and H···O separations of 1.004 and 1.604 Å (Δ = 0.6 Å), which are again typical of medium-strength hydrogen bonds. This geometry is in strong disagreement with the experimentally determined short hydrogen bond in UPA, with values at 15 K of O–H, 1.158(3) Å, H···O, 1.267(3) Å, and O···O separation of 2.420(2) Å; these isolated molecule calculations, therefore, do not adequately describe the short hydrogen bond in this system.
In order to investigate this complex further, and in particular to investigate the intermolecular HB interaction, a set of plane-wave (periodic) DFT calculations were also carried out on the UPA complex. By incorporating the periodic nature of the crystal lattice into the calculations, it is hoped to reproduce the molecular structure in the crystal more accurately. These calculations were conspicuously successful—for the intermolecular HB in the complex, plane-wave DFT predicts a short, strong HB with O···O separation of 2.42 Å, and an asymmetry of Δ = 0.22 Å, reflecting the significant elongation of the O–H (1.105 Å) and shortening of H···O (1.329 Å) distances. While still representing a greater asymmetry than that found in the crystal structure (Δ = 0.11 Å), the plane-wave DFT calculations clearly do give a better description of the known structure in the crystal environment, and consequently the deductions from these calculations may yield useful insight into the nature and behaviour of the HB potential. In a second study we have taken this work further, by attempting to reproduce the thermally-induced proton migration [38]. Plane-wave DFT Molecular Dynamics techniques have been developed to apply to this and related problems emerging from our structural work, and these have been applied to urea–phosphoric acid. The advantage of this method is that we are no longer constrained to carry out ‘0-K’ calculations, but can provide the system with more energy and hence simulate the effect of increasing temperature. The results of these calculations have shown great promise in allowing the temperature dependence of the system to be modelled, offering possible explanations for the apparent proton migration, including the possibility that the shape of the hydrogen bond potential is indeed evolving subtly with temperature in the crystal lattice environment [38].
We have also carried out plane-wave DFT calculations on potassium hydrogen maleate [29]; indeed, the findings of the calculations carried out were perhaps the most significant aspect of this study. For the isolated hydrogen maleate anion, the calculations support an asymmetric hydrogen bond as the lowest energy structure (in strong disagreement with the situation found in the solid state). This clearly mirrors the situation found for urea–phosphoric acid, and again shows the isolated molecule calculations to be not well suited to studies of hydrogen bonds such as those of interest here. Once again moving into the periodic environment gives significant benefits; for the potassium hydrogen maleate crystal, the periodic calculations show clear evidence of a flat potential energy surface, and consequently favour the symmetric intramolecular hydrogen bond, even when the crystallographic symmetry is removed from the calculations [29]. Thus, by the use of ab initio molecular orbital calculations and plane-wave DFT calculations, this demonstrates that the nature of the intramolecular hydrogen bond potential for the hydrogen maleate anion changes as a consequence of state. In the gas phase (i.e. isolated molecule) an asymmetric (Cs) hydrogen bond is the lower energy arrangement, but in a crystalline environment with potassium counter-cations the potential energy well flattens to favour the symmetric (C2v) bonding environment.
5 Summary and conclusions
A range of experimental and computational techniques has been used to study a series of short hydrogen bonds in a range of molecular materials. The potential of modern single crystal neutron diffraction using large area detector arrays (offering > 2π Sr) solid angle coverage is emphasised by this work. Dramatically reduced data collection times together with the availability of flexible sample environment on such instruments has allowed us successfully to survey temperature–pressure space in several materials and to build up a fuller picture of the structural evolution in these. The full description provided by neutron diffraction is also supported by advanced X-ray diffraction methods employing Fourier imaging as an important diagnostic of hydrogen atom behaviour, along with the fundamental insights being afforded by complementary computational methods. The importance of carrying out calculations in a periodic environment has been stressed by the results found here; even high-level isolated molecular calculations are found to be inadequate in describing hydrogen-bonded systems. In contrast, plane-wave DFT methods offer a more realistic description of the systems studied, in agreement with experiment, and hence will allow the basis of these hydrogen bonds to be probed.
Acknowledgements
This work was supported by EPSRC under grant GR/R04690. The authors thank their collaborators on the range of published work discussed here, including A.J. Florence, A.E. Goeta, S.M. Harte, C.A. Morrison, A. Parkin, K. Shankland and N. Shankland, and also colleagues on the SXD instrument, M.J. Gutmann and D.A. Keen.