

1 Introduction
The spin–spin coupling analysis of 1D NMR spectra describes how nuclei are scalarly coupled within molecules. Scalar couplings are important NMR parameters that provide constraints for the building of 3-D molecular structures [1]. The high magnetic fields at which NMR is performed nowadays reduce the probability of strongly coupled homonuclear spin systems, so that first-order analysis is most often sufficient for measuring coupling constants. Each multiplet can therefore be analyzed independently of the others within the same spin system. The automatic extraction of coupling constants is a problem to which solutions have already been proposed ([2–11], and references cited therein). Human interpretation of a multiplet structure relies on the recognition of elementary peak cluster shapes [12]. Automated methods for first order multiplet analysis mimic this process [11].
A method based on time-domain analysis has already been described by our group [13,14]. Its basic principle was later extended to increase its performance. This communication describes the process that is presently implemented in the AUJ (automatic J couplings) computer software (www.univ-reims.fr/LSD/JmnSoft/Auj).
AUJ is implemented as a GIFA [15] macro command that performs some pre-processing and calls a binary program whose source file is written in C language. The latter uses a library that contains the truely active part of the AUJ algorithm and that is designed to be invoked from any type of NMR processing software. The library uses the time-domain data as input and produces a set of coupling constants as well as the corresponding reconstructed time-domain signal.
2 Process outline
Analysis starts with data point extraction of the user-selected multiplet. The multiplet is then converted to complex time-domain data by inverse Fourier transformation. Alternatively, a column of a 2-D J-resolved spectrum can also be simply extracted by the user and back-converted to real time-domain data. The AUJ algorithm is able to handle both input types. Complex data is supposed to originate from a non-centered non-symmetrical multiplet and therefore must be transformed to real data (step 1). The ‘log-abs’ algorithm [13,14] is then used (step 2) to produce a 1-D J-spectrum that is then optimized (step 3) and analyzed to produce a first set of coupling constants and multiplicities (step 4). At this stage, J values can be accurate but multiplicities are usually not. The latter are optimized (step 5) and the resulting values used to refine the final J values (step 6). Finally, the centered multiplet (when input data is complex), the reconstructed time-domain data, and the J values are exported back to the calling GIFA macro command and made visible through its graphical user interface, so that an algorithm failure is immediately visible. The ratio of the spectral noise level with the root-mean-square deviation between original and reconstructed multiplets is also a good performance indicator. The analysis process depends on empirically adjusted parameters whose default values, provided in the following paragraphs, can easily be adjusted by means of a single control panel. In practice, the proposed defaut parameter values fit well with most of the situations that were tested by the authors.
2.1 Step 1: Multiplet centering and symmetrization
Time-domain data is first zero-filled (16 times by default) to obtain a high resolution multiplet by Fourier transformation. Then, a pivot frequency is searched so that frequency reversal around the pivot point leads to a spectrum that looks, at best, identical to the original one. Reversal and comparison are not directly performed on the high-resolution spectrum but on a binary-valued version of it that is built as follows. The real part of the spectrum is first divided by its Euclidian norm. Each value in the normalized spectrum is replaced by 1 if its value is greater than a given threshold (0.03 by default) and, if not, replaced by 0. The pivot position is selected so that the number of ones is maximum in the point-by-point product of the binary spectrum by its reversed version. The optimum pivot frequency value is used to frequency shift the original time-domain signal by multiplying it by the adequate linear phase ramp function. Frequency symmetrization is achieved de facto by setting the imaginary part of the resulting time-domain data to zero. Centering failure may occur for strongly unsymmetrical multiplets and threshold adjustment may be necessary.
2.2 Step 2: ‘log-abs’ analysis
A first-order multiplet is described in the time domain by:
| (1) |
| (2) |
The model described by equations (1) and (2) is slightly different from the usual one, because it imposes N (80 by default) predefined J values:
| (3) |
| (4) |
Considering that
| (5) |
2.3 Step 3: Optimization of the 1-D J-spectrum
The crude 1-D J-spectum does not exploit all available data and is strongly affected by noise. Its refinement is possible through the minimization of the least squares residue R:
| (6) |
2.4 Step 4: Analysis of the refined 1-D J-spectrum
Each series of contiguous
| (7) |
| (8) |
2.5 Step 5: Multiplicity and
This step is a grid search of the best integer multiplicities and
| (9) |
2.6 Step 6: Jk optimization
The low resolution in the 1-D J-spectrum may lead to believe that two different J values are identical. Therefore, the multiplet is considered as being produced by the effect of K independent couplings with:
| (10) |
Each Jk value is perturbed by addition of a small deviation drawn from a random number generator (±0.05 Hz by default). The Jk and
3 Results
The proton NMR spectrum of sucrose in DMSO-d6 was recorded at 500 MHz. The quadruplet-like signal at δ = 3.81 ppm (Fig. 1, left) is obviously more complex than a regular quadruplet. This particular multiplet would clearly be a difficult problem to solve for any method based on peak list analysis.
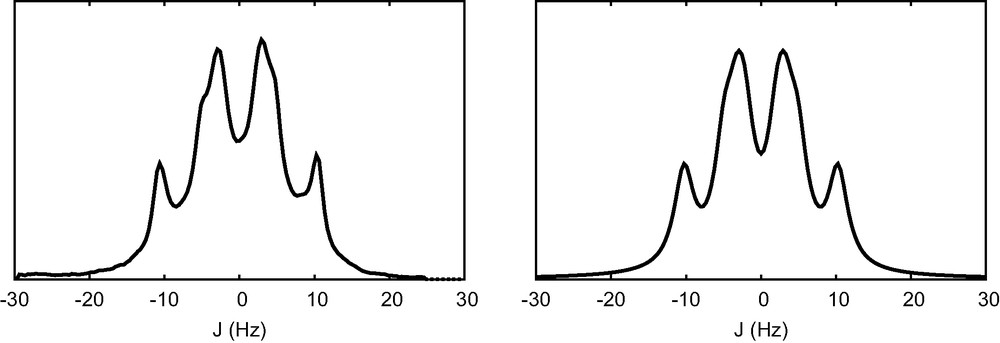
The multiplet at δ = 3.81 ppm (TMS as reference) in the 1H NMR spectrum of sucrose dissolved in perdeuterated DMSO (left). The multiplet that is reconstructed from the AUJ analysis, with J = 5.37 Hz, 7.04 Hz, 8.14 Hz and
The 1-D J-spectrum is analyzed as J = 1.47 Hz (0.55), 2.13 Hz (0.54), 5.57 Hz (1.32), 7.20 Hz (0.72), and 8.25 Hz (0.80), where numbers in parenthesis are the
A more difficult problem was given to AUJ, the analysis of a multiplet that is simulated on the basis of results presented in [11] for the methine proton of 3-bromo-2-methyl-1-propanol (Fig. 2, left). The multiplet is a quadruplet of quintet with J = 5.43 Hz and 6.76 Hz, respectively. Some computer generated noise is added to the spectrum and the line width is chosen so that peak clusters are poorly resolved. The 1-D J-spectrum is interpreted as J = 1.10 Hz (0.77), 5.46 Hz (1.24), and 6.69 Hz (1.80). Again, the noise introduces unwanted small coupling constants in the 1-D J-spectrum. The multiplicity optimization step finds the correct result and the final refinement produces J1 = 5.39 Hz, J2 = 5.40 Hz, J3 = 5.41 Hz, J4 = J5 = J6 = 6.78 Hz, J7 = 6,79 Hz, a result that compares well to what is expected.
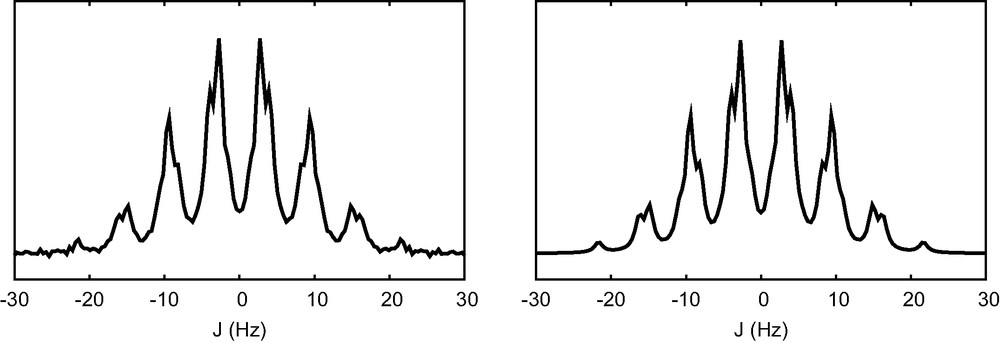
Experiment on simulated data, using parameters published in [11] for the methine proton of 3-bromo-2 methyl-1-propanol. Simulated spectrum, using J1 = J2 = J3 = 5.43 Hz, J4 = J5 = J6 = J7 = 6.76 Hz and
4 Conclusion
This Communication shows that the AUJ algorithm provides a pertinent way to analyze complex multiplets. The modeling of time-domain data ensures a reliable result on poorly resolved signals, even though non-ideal lineshapes and high noise levels may lead the user to modify the default algorithm parameters. However, it should always been remembered that the best first order analysis algorithm ever written cannot provide a safe and useful result if carried out on a part of a strongly coupled spin system. Further development of AUJ will deal with parameter selection improvement, the interfacing of the algorithm with commercial NMR data processing software, as well as its extension to slices of 2-D NMR spectra, different from J-resolved ones, in order to analyze nearly or fully superimposed multiplets.
Acknowledgements
We thank Dr. Karen Plé for linguistic improvement.
Vous devez vous connecter pour continuer.
S'authentifier