

1 Introduction
Since Honda and Fujishima [1] first reported H2 generation from water via photocatalysis with TiO2, numerous studies have examined transition metal oxide semiconductors as candidate water splitting photocatalysts [2]. However, the band gaps of most oxide semiconductors, including TiO2, are such that they are active only under ultraviolet (UV) light. Thus, for more efficient use of solar energy, there is an urgent need for photocatalysts that can generate hydrogen under visible light. Recently it was reported that Ni-doped InTaO4 could evolve H2 from pure water under visible light, albeit with very low activity [3]. Several recent studies have investigated the electronic structures of InTaO4 and N-doped InTaO4 [4,5]. To our knowledge, however, the systematic studies on the electronic structures of InTaO4 and the absorption of visible light, has yet to be done.
In this report, we present the results of electronic structure calculations on pristine InTaO4, Ni-doped and A-doped (A = C,N,S) InTaO4, and InTaO4 containing oxygen vacancies. The calculated results for InTaO4 were used to clarify the band gap narrowing observed experimentally by Zou et al. [3]. The relationship between the band gaps in each system and the absorption of visible light is discussed.
2 Computational details
InTaO4 has a wolframite structure that belongs to the space group P2/a. This structure is made up of two types of octahedron, InO6 and TaO6, which form a layered structure by sharing corners [4,5]. InTaO4 contains two kinds of oxygen: O(1), which is coordinated with two In atoms and one Ta atom; and O(2), which is coordinated with two Ta atoms and one In atom. In the calculations, a 2 × 2 × 1 supercell was used in constructing the oxygen vacancy and Ni-doped structures. Thus one supercell consists of eight indium, eight tantalum, and 32 oxygen [16 O(1) and 16 O(2)] atoms. To model the structure of InTaO4 with oxygen vacancies, one oxygen ion (O2–) of the 16 O(1)s (or 16 O(2)s) was removed from a 2 × 2 × 1 supercell in the periodic structure. For the Ni-doped model, one of the eight indium ions (In3+), in a supercell was replaced with a (Ni2+) ion. This corresponds to a Ni content of x = 0.125 in In1–xNixTaO4, which falls within the experimental Ni content range of x = 0.0–0.2 reported by Zou et al. [3]. For anion (A)-doped case, one of 16 O(1)s or 16 O(2)s is replaced by one anion of C, N, or S.
All electronic structure calculations were carried out using the Vienna ab-initio simulation package (VASP) [6,7], which is based on density functional theory. The wavefunctions of the valence electrons were expanded using a plane wave basis set with a cutoff energy of 380 eV. The core electrons were treated using ultrasoft pseudopotentials [8]. The exchange and correlation interactions were modeled using the generalized gradient approximation (GGA). All calculations presented here are spin-unpolarized, except Ni-doped case where total number of electrons of the system is odd number.
3 Results and discussions
First, we optimized the atomic positions of pristine InTaO4. The calculated lattice parameters and atomic coordinates were very similar to those reported previously [4]. The atomic positions for the oxygen vacancy structure, Ni-doped, and anion(A)-doped structure were obtained by relaxing structures constructed using the lattice parameters calculated for the pristine InTaO4. After introducing an oxygen vacancy, the bond lengths between the cations and oxygens nearest to the vacancy site changed by 0.01–0.69 Å. However, the positions of atoms at the second nearest neighbor sites relative to the vacancy site hardly changed. Substitution of In3+ with Ni2+ induced no noticeable structural changes, except that the Ni–O bond length was shorter than the original In–O bond length due to the smaller ionic radius of Ni2+ compared to In3+. These findings agree with a previous report that doping of InTaO4 with a Ni content of x = 0.15 induced a volume reduction of only 0.26% [3]. Substitution of O with N induced no noticeable structural changes, while substitution of C and S induced some structural changes near the substituted site. The bond lengths between the cations and the substituted anion changed by 0.04–0.43 Å. We have substituted O(1) and O(2) with an anion, respectively. The energy differences between O(1) substitution and O(2) substitution with C, N, and S are 0.29 eV, 0.05 eV, and 0.02 eV, respectively. For anion-doped cases, since the energy differences are small and the electronic structures are very similar for O(1) and O(2) substitutions, here we presented the calculated results for O(1) substitution only.
We calculated the density of states (DOS) for pristine InTaO4, InTaO4 with O(1) or O(2) vacancies, and InTaO4 doped with Ni, C, N, and S. Fig. 1 shows the total DOS (TDOS) for pristine and oxygen vacancy states. The calculated band gap between the valence band (VB) and conduction band (CB) for the pristine structure (Fig. 1a) is 3.46 eV, which is slightly lower than the previously reported value of 3.7 eV [4,5]. It is plausible considering that they [4,5] used different computational methodologies. As expected, we found that the VB and CB consist predominantly of O 2p and Ta 5d, respectively.
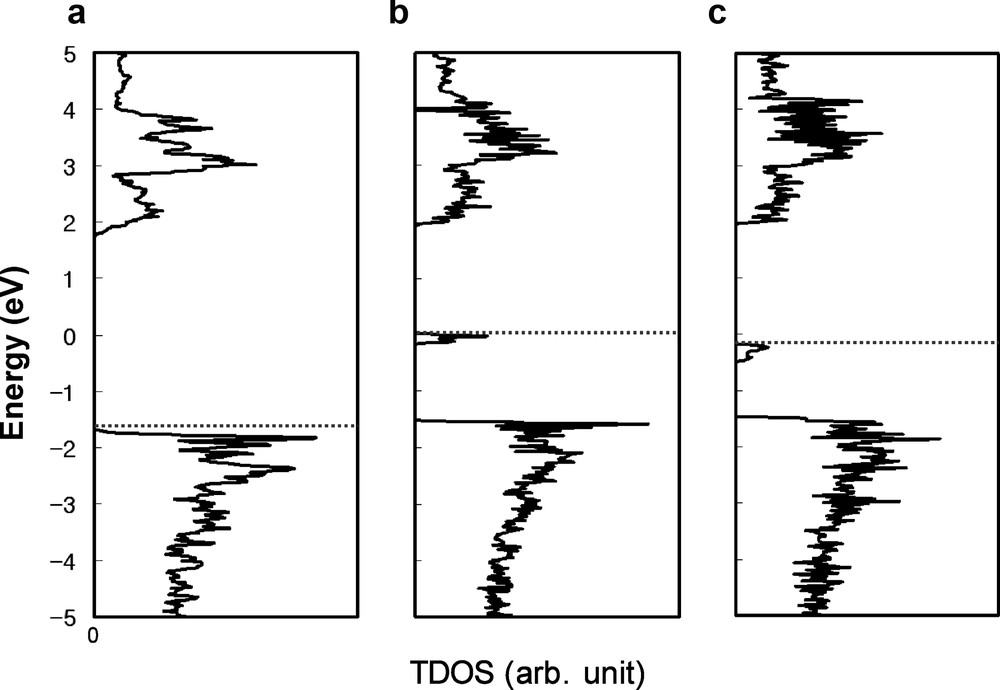
The calculated total density of states (TDOS) for (a) pristine structure, (b) O(1) vacancy structure, and (c) O(2) vacancy structure. HOMO level (the energy for the highest occupied states) is represented with dashed line for each case.
Previous band gap assignments based on experimental optical absorption data are inconsistent, as shown in Table 1. Zou et al. [3], assigned the 2.6 eV onset to the bandgap between the VB and the CB for undoped InTaO4, whereas Matsushima et al. [5] assigned this bandgap to the 4.3 eV onset and suggested that the 2.6 eV onset is due to the presence of gap states between the VB and CB.
| absorption onset (eV) | Zou et al.3 | Matsushima et al.5 | This work |
| 4.3a | – | VB → CB | VB → CB |
| 2.6a,b | VB → CB | GS → CB | GS → CB |
| 2.3b | DS → CB | – | DS → CB |
When compared with the experimental observations, the band gap calculated in the present work (3.46 eV) is larger than the 2.6 eV onset, but smaller than the 4.3 eV onset. Given that density functional theory usually underestimates band gaps because of the discontinuity in the exchange-correlation potential, we assign our calculated band gap to the 4.3 eV onset rather than the 2.6 eV onset, in agreement with Matsushima et al. [5] The source of the 2.6 eV onset of InTaO4 observed experimentally [3] can be explained from the results of our oxygen vacancy calculations, which revealed new states between the VB and CB (see Fig. 1b and c). HOMO level (the energy for the highest occupied states) is represented with dashed line in the figures. These new gap states are below the HOMO level, and hence are all occupied states. By carrying out a partial DOS analysis, we established that the dominant contribution to these gap states derives from the electronic states of the atoms nearest to the vacant site. The major contributions were found to be from In 5s and 5p, hybridized with some Ta 5d and O 2p. Oxygen vacancy induces dangling bonds on the nearest cations, which generates gap states in the middle of the pristine band gap. The transition from these gap states to the CB is consistent with the observed 2.6 eV onset, as listed in Table 1. These findings imply that the undoped sample used in the experiments contained oxygen vacancies, a common characteristic of transition metal oxide samples. Our calculations thus clarify that the gap states are defect states induced by oxygen vacancies, thereby resolving the conflicting assignments reported previously [3,5]. Because the energy difference between the gap states and the CB is in the energy region of visible light, the electrons of the gap states can easily be excited to the CB by absorption of visible light. Moreover, electrons in the VB can be excited to the gap states by visible light, and then subsequently excited to the CB by further absorption of visible light. These transitions make possible photocatalytic water splitting under visible light, even with undoped InTaO4 [9].
The calculated TDOS of the Ni-doped structure, shown in Fig. 2, exhibits new states within the band gap of the pristine structure. Additional states also appear near the top of the pristine VB. By partial DOS analysis, we found that these new gap states are predominantly unoccupied Ni 3d states, whereas the additional states near the top of the VB are predominantly occupied Ni 3d states. However there exists some hybridization between the Ni 3d states and the O 2p and Ta 5d states of nearby atoms.
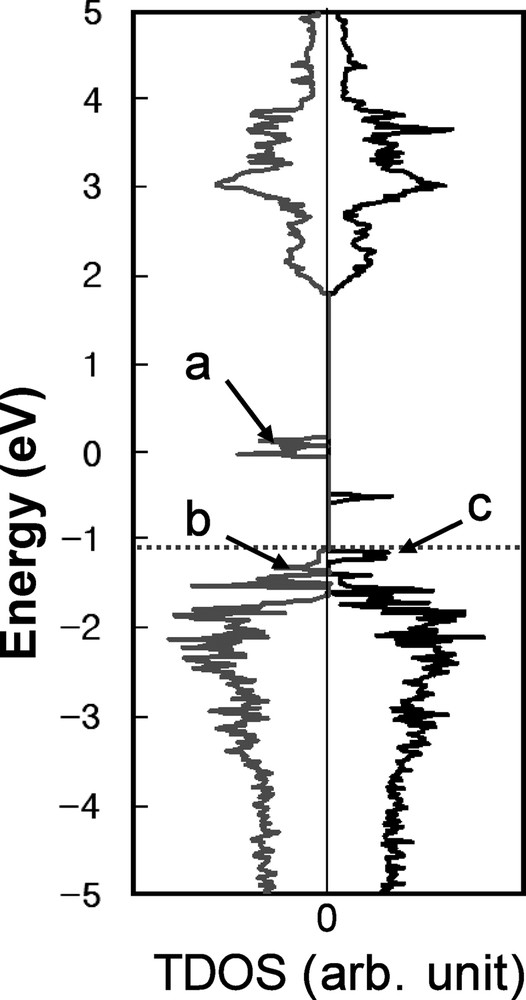
The calculated total density of states (TDOS) for Ni-doped structure. The dark line is for spin up states and the gray line is for spin down states. HOMO level is represented with dashed line.
Comparing TDOS of Ni-doping (Fig. 2) and oxygen vacancy states (Fig. 1), the unoccupied Ni-doping states (marked ‘a’ in Fig. 2) are located near the occupied defect gap states within the pristine band gap. The electrons in the defect gap states can be easily excited to the unoccupied doping states, because the energy difference between them is small enough for thermal excitation. Then the electrons in the Ni-doping states (a) can be subsequently excited to the CB states. Hybridization of Ni 3d, O 2p and Ta 5d in the doping states can enhance these subsequent excitations, as mentioned above. This clearly explains why the band gap narrowed from 2.6 to 2.3 eV on doping Ni into the InTaO4 structure [3], as shown in Table 1. Comparing Fig. 2 and Fig. 1a, the occupied Ni-doping states locate immediately above the pristine VB (marked ‘c’ and ‘d’ in Fig. 2), which also serve to reduce the pristine band gap. This predicted behavior is borne out in the experimental data reported by Zou et al., which shows that Ni-doping caused the onset of optical absorption near 4.3 eV to shift to 3.8 eV; however this obvious evidence that Ni-doping leads to the formation of additional states on the top of the VB was not pointed out previously [3].
For anion-doped cases, the calculated TDOS are shown in Fig. 3. As shown in Fig. 3a, C-doping induced new occupied gap states, which is similar to the gap states induced by oxygen vacancy shown in Fig. 1. From partial DOS analysis, we have found that these gap states consist of C 2p predominantly with little O 2p hybridization. N-doping and S-doping also induced the gap states as shown in Fig. 3b and c. However, these gap states are close to the VB top, which can narrow the band gap. These results agreed with the previous study of N-dope InTaO4 [5]. From partial DOS analysis, the gap states of N-doping (S-doping) consist of mainly N 2p (S 3p) states with little O 2p hybridization.
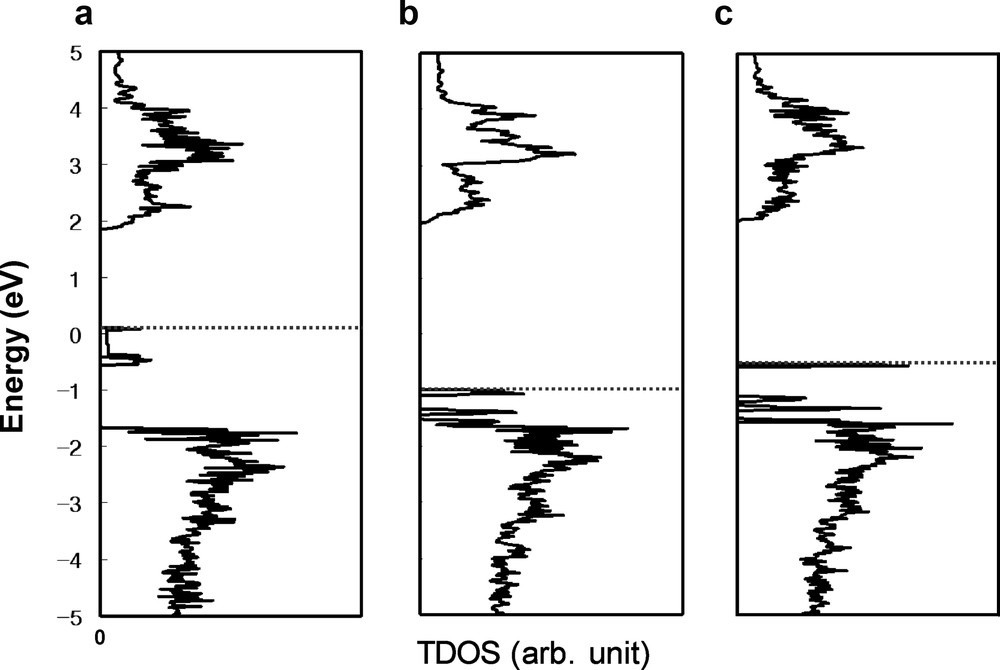
The calculated total density of states (TDOS) for (a) C-doped, (b) N-doped, and (c) S-doped structure. HOMO level is represented with dashed line for each case.
4 Conclusions
From our calculations, we have established that undoped InTaO4 can absorb visible light via gap states induced by oxygen vacancies. In addition, the calculated DOS for Ni-doped InTaO4 revealed additional states immediately above the gap states and on top of the VB states. These new doping states not only reduce the defect band gap due to oxygen vacancies but also the band gap between the VB and CB. Our calculations clearly show that Ni-doping can induce band gap narrowing. The appearance of gap states in the DOS of InTaO4 containing oxygen vacancies or doped with Ni indicates two key avenues for producing InTaO4 capable of absorbing visible light, which was shown in the previous experiment [3]. For anion-doped cases, we have found that C-doping induced different electronic states from N-doping and S-doping. C-doping generates the isolated gap states in the middle of the pristine band gap, while N-doping (S-doping) generates the gap states above the top of the pristine VB, which induces band gap narrowing. The C-doping gap states seem very similar to the defect gap states induced by oxygen vacancy. From our calculated results, we would suggest the experimental treatment to induce oxygen vacancies or C-doping in InTaO4 to generate more gap states for efficient absorption of visible light.
Acknowledgements
This work was supported by the Ministry of Science and Technology (MOST) of Korea through the Hydrogen Energy R&D Center and the National R & D Project for Nano Science and Technology.