

1 Introduction
The artificial photosynthesis of a fuel like methanol from CO2 and H2O is a key challenge of the generation of renewable energy. For heterogeneous systems, the reaction has first been reported for large bandgap semiconductor materials such as TiO2, SrTiO3, ZnO, or SiC upon irradiation with UV light [1–4]. Methanol and methane were detected after prolonged irradiation along with CO, formic acid and O2 as minor products. Use of smaller bandgap materials like ZnSe or CdSe afforded photoreduction of CO2 with visible instead of UV light [5–9]. However, organic donors or other sacrificial reagents were required for sustaining the reaction, preventing thus far the development of a concept for visible light-induced CO2 reduction by H2O based on this approach. Anpo and coworkers have recently found that ligand-to-metal charge-transfer (LMCT) excitation of isolated Ti centers of framework substituted micro or mesoporous silicates by UV light affords the reduction of CO2 by H2O at substantially better efficiency than dense phase TiO2 particles [10–13]. Hence, the single-center version of the metal oxide photocatalyst approach offers a promising direction, but no results have been reported so far that would allow the utilization of visible instead of UV photons.
We report here on visible light-activation of Ti or Zr centers in a silicate molecular sieve by engaging a second metal center as donor, linked to the group IVB metal by an oxo bridge. Oxo-bridged metal-to-metal charge-transfer (MMCT) moieties are expected to absorb visible and even near-infrared light based on spectroscopic work on solid mixed metal oxides [14]. The great flexibility in the selection of metal and oxidation state might open up access to solar light-absorbing chromophores with a wide range of redox properties. For exploring MMCT absorptions of binuclear units covalently anchored in mesoporous silicates, Ti or Zr linked to d10 or s2 metals like Cu(I) or Sn(II) offer a convenient starting point because they lack ligand-field or low LMCT bands that otherwise might complicate the spectra in the visible region. A silicate sieve featuring Zr–O–Cu(I) sites was found to split CO2 to CO upon excitation of the MMCT chromophore.
2 Experimental
MCM-41 sieve was synthesized according to the procedure described by Lin et al. [15]. Grafting of Ti or Zr centers was accomplished using the corresponding dicyclopentadienyl dichloride precursor [16]. Tetrahedral coordination of Ti in Ti–MCM-41 was confirmed by XANES and UV diffuse reflectance spectroscopy [17]. ICP-AES analysis gave ratios Si/Ti = 54 and Si/Zr = 24. For the assembly of TiCu(I) (ZrCu(I)) moieties, 0.2 g of Ti–MCM-41 (Zr–MCM-41) crystallites were added to a solution of Cu(I)(NCCH3)4PF6 in 100 ml CH2Cl2 (0.1 wt%) inside a N2 glove box. The mixture was stirred at 40 °C for 30 min, filtered and washed (Ti/Cu = 0.82, Zr/Cu = 1.3). Monometallic Cu(I)–MCM-41 was prepared by the same procedure using the neat silicate as starting material (Si/Cu = 64).
As synthesized material was heated to 300 °C for 12 h under vacuum in order to remove residual CH3CN, which was verified by FT-IR spectroscopy. 18O Labeled Zr–MCM-41 was prepared by heating as-synthesized Zr–MCM-41 (which features a residual cyclopentadienyl ligand on each grafted Zr center) in an 18O2 atmosphere at 550 °C, resulting in the formation of Zr–18OH sites as well as exchanged Si–18OH groups. Preparation of TiSn(II) groups was conducted by exposing a SnCl2 solution in acetonitrile (0.1wt%) to Ti–MCM-41 powder (Ti/Sn = 1.92). XRD measurements (Siemens model D500 Cu Kα) of the bimetallic sieves showed no loss of long-range order upon grafting.
UV–Visible DRS measurements were performed on Shimadzu model UV-2100 spectrometer equipped with an integrating sphere model ISR-260. Pressed wafers of the bimetallic MCM-41 sieves (10 mg) were loaded into an optical vacuum cell under N2 atmosphere. While mounted on the integrating sphere, the cell was connected to a vacuum manifold for evacuation or loading of oxygen gas. The same procedure was used for loading of the sample into a transmission infrared vacuum cell equipped with CaF2 or KBr windows. FT-IR spectrometers used were Bruker model IFS88 or IFS66V equipped with LN2 cooled MCT detectors with 8 or 25 µm bandgap. Raman spectra were recorded with an FT-Raman module Bruker model FRA 106/S equipped with a LN2 cooled Ge detector. For visible light irradiation, the emission of a W-source was used. Photolysis at 355 nm was conducted with the third harmonic emission of a Quanta-Ray Nd:YAG laser model DCR-2A.
3 Results and discussion
3.1 Anchored MMCT chromophores for visible-light activation of Ti or Zr centers
Addition of Ti–MCM-41 crystallites to a methylene chloride solution of Cu(I)(NCCH3)4PF6 resulted in instant yellow coloration of the silicate sieve. No such color change was observed when using neat MCM-41, indicating that the visible absorption requires the presence of both Cu(I) and Ti. The DRS of a pressed wafer of TiCu(I)–MCM-41 under vacuum shown in Fig. 1a, features an absorption tail extending from the UV to about 600 nm. As can be seen from the Cu–MCM-41 spectrum of the same Figure, the new absorption of the bimetallic material far exceeds the Cu(I) absorption. We attribute the broad continuous band shown in the difference spectrum (insert of Fig. 1a) to the Ti(IV)/Cu(I)→Ti(III)/Cu(II) MMCT transition. This is supported by the observed decrease of the band in the 300–600 nm region upon exposure of the TiCu(I)–MCM-41 material to 1 atm of O2 gas at room temperature under concurrent growth of the Cu(II) d–d band around 800 nm [18], shown in Fig. 1b. Oxidation of Cu(I) is expected to result in a loss of MMCT absorption intensity. An analogous absorption in the range 250–500 nm exhibiting the same O2 sensitivity was found for ZrCu(I)–MCM-41 sieve and is assigned to the Zr(IV)/Cu(I)→Zr(III)/Cu(II) MMCT transition. Fig. 2a shows that there is an additional optical absorption for the bimetallic ZrCu(I)–MCM-41 material when compared with the monometallic Cu(I)–MCM-41 sieve, while Fig. 2b displays the depletion of the MMCT band and growth of the Cu(II) absorption upon O2 exposure. Similarly, grafting of Sn(II) onto Ti–MCM-41 gave a sieve whose optical spectrum is shown in Fig. 3. The bimetallic material has an intense absorption in the blue spectral region that is absent in monometallic Sn(II)–MCM-41. The only assignment we can conceive of is Ti(IV)/Sn(II)→Ti(III)/Sn(III) MMCT. The relatively short wavelength onset of the charge-transfer transition reflects the instability (high reduction potential) of Sn(III).

(a) Diffuse reflectance spectra of TiCu(I)–MCM-41 and Cu(I)–MCM-41. The insert shows the difference of the two spectra. (b) DRS before and after exposure of TiCu(I)–MCM-41 to air at RT.
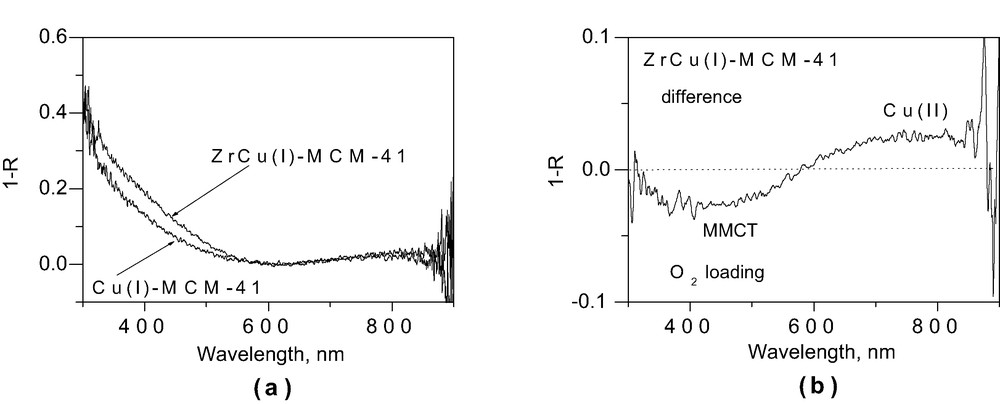
(a) UV–Vis DRS of ZrCu(I)–MCM-41 and Cu(I)–MCM-41. (b) Difference DRS spectra recorded after and before exposure of ZrCu(I)–MCM-4 to O2 gas (1 atm) at RT for 8 h.

UV–Vis DRS of TiSn(II)–MCM-41 after heating under vacuum. The insert shows the difference of TiSn(II) and Sn(II) spectra.
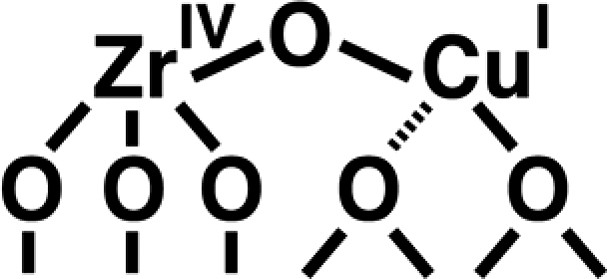
The key information on the structure of the bimetallic sites was furnished by the low-frequency metal–oxygen stretching modes. The FT-IR spectrum of TiCu(I) and ZrCu(I) sieve show a Cu(I)–O stretch absorption around 650 cm–1. When grafting the Cu(I) precursor onto Zr–MCM-41 featuring Zr–18OH and Si–18OH groups, the band showed a red shift of 30 cm–1 characteristic for a Cu–O mode. Further support for this assignment came from the observation of the same band when grafting the Cu(I)(NCCH3)4+ complex onto porous TiO2 or ZrO2 (band maxima around 640 cm–1). We conclude that ν(Cu–O) of Ti–O–Cu(I) and Zr–O–Cu(I) absorb at 640 cm–1. Upon exposure of the bimetallic sieve to an O2 atmosphere, the ν(Cu(I)–O) absorption decreased under concurrent growth of the Cu(II)–O absorption at 560 cm–1 (TiCu–MCM-41, shown in Fig. 4) or 540 cm–1 (ZrCu–MCM-41). The band agrees well with the Cu(II)–O modes reported for Cu(II) complexes [19], CuO films [20], or basic Cu(II) salts [21]. In the case of TiSn(II)–MCM-41, the Sn(II)–O infrared absorption was observed at 610 cm–1, and shifts to 595 cm–1 upon oxidation of the material by exposure to O2 gas. These infrared observations strongly support the assignment of the visible absorptions to MMCT chromophores and indicate that the anchored binuclear moieties are oxo-bridged (Scheme 1). In addition to the Cu–O stretching mode, all three bimetallic sieves show a band at 930 cm–1 that is typical for Si–O stretch vibrations perturbed by the presence of a metal center. The position of this band is quite insensitive to the nature of the metal [22,23], and its intensity is substantially higher for the bimetallic compared to the monometallic materials, as expected.

(a) FT-IR spectrum of evacuated TiCu(I)–MCM-41 sieve. (b) Spectrum after exposure to air. For background subtraction, the spectrum of neat MCM-41 was used.
Irradiation of the MMCT chromophore of the evacuated TiSn(II)–MCM-41 powder (77 K) with 355 nm light at 160 mW cm–2 for 30 min. led to the g = 1.907 EPR signal of Ti3+ shown in Fig. 5. The same signal was observed upon shining visible light of a filtered W-source (λ > 400 nm) for 30 min, also shown in Fig. 5. The signal was identical to that observed for an unperturbed Ti3+ (e.g., by 266-nm irradiation of Ti–MCM-41), consistent with the spontaneous conversion of unstable paramagnetic Sn(III) to EPR-silent Sn(IV). No EPR signal was detected upon visible light (or 355 nm) irradiation of MCM-41 sieve that contained only grafted Sn(II) or Ti. We conclude that excitation of the Ti(IV)-O-Sn(II) moiety leads to activation of Ti under visible light. The longevity of the charge separation is attributed to the build up of Ti3+ by trapping of electrons released spontaneously by the unstable Sn(III) generated by MMCT excitation.
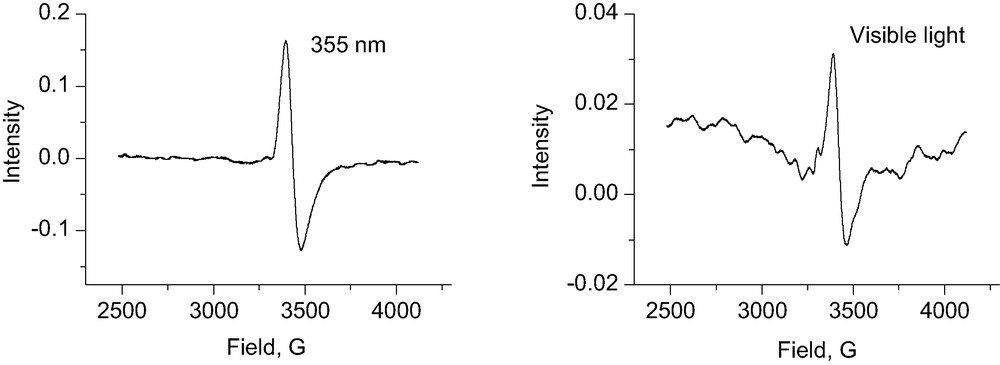
EPR spectra of TiSn(II)–MCM-41 irradiated with visible light (> 400 nm) (right) and 355 nm radiation (left) at 77 K for 30 min. EPR spectra were recorded at 20 K.
3.2 Photoreduction of CO2 in ZrCu(I)–MCM-41 sieve
Loading of 750 Torr CO2 gas into a pressed water of ZrCu(I)–MCM-41 and irradiation of the MMCT chromophore at 355 nm at low power (110 mW cm–2) resulted in infrared growth at 2148 and at 1600 cm–1, as shown in the top trace of Fig. 6. The broad 2148 cm–1 band originates from CO product bound to Cu(I), as confirmed by adsorption of an authentic sample of CO gas. This is further confirmed by the observation of a 47 cm–1 isotope shift of the band when using 13CO2 (Fig. 6a, bottom trace). The 1600 cm–1 band does not exhibit any shift when using 13CO2 as reactant; therefore, it can unambiguously be assigned to H2O. On the other hand, photoreduction of C18O2 led to growth of H218O at 1594 cm–1 (Fig. 6b, bottom trace), indicating that the oxygen atom of water stems from CO2. The C18O product absorbed at 2096 cm–1. No growth of isotopically labeled CO was detected when using a ZrCu–MCM-41 sieve with Cu in the oxidized Cu(II) state. The results prove that the carbon monoxide originates from the MMCT-induced photoreduction of CO2. No significant CO growth was detected when attempting CO2 photoreduction in TiCu(I)–MCM-41, whose MMCT absorption is much more intense (see Figs. 1 and 2). This is consistent with the much lower reducing power of transient Ti3+ compared to Zr3+.
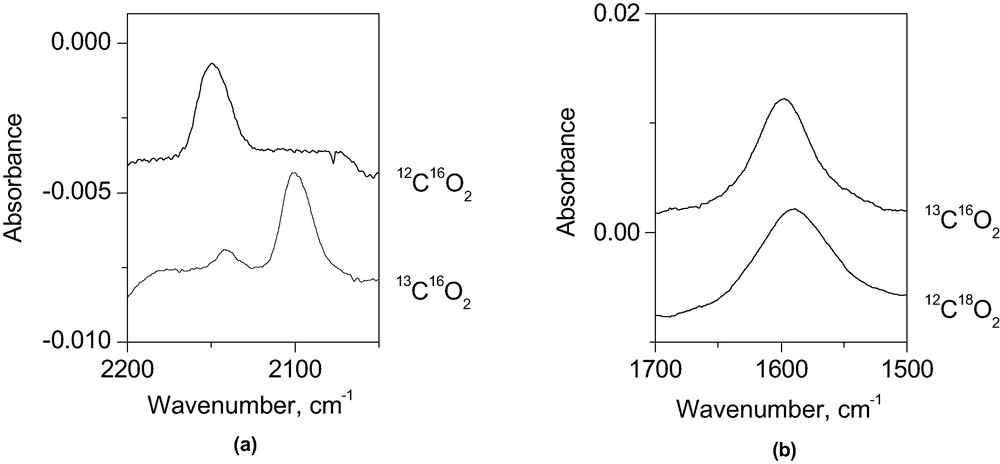
FT-IR spectra recorded before and after 355-nm irradiation of ZrCu(I)–MCM-41 loaded with 1 atm of CO2, 13CO2, or C18O2 at 355 nm (110 mW cm–2) for 2.5 h.
As expected, photolysis induced concurrent intensity loss in the Cu(I)–O stretching region, with the maximum bleach occurring at 643 cm–1, as shown in Fig. 7. This confirms that the CO2 photoreduction is accompanied by oxidation of Cu(I) linked to the Zr center. Interestingly, comparison with the depletion spectrum upon oxidation by O2 shows that photolysis results in the selective loss of a shoulder at 643 cm–1 whereas the whole ν(Cu(I)–O) absorption profile decreases upon chemical oxidation of Cu(I). Since MMCT-induced photolysis results only in the oxidation of Cu(I) centers that are part of a binuclear site, this indicates that the 643 cm–1 component of ν(Cu(I)–O) originates from a Cu(I)–O–Zr mode. It constitutes our most direct evidence yet for the oxo-bridged nature of the MMCT site. It is worth adding that the assignment is based on several other control experiments. This includes the absence of depletion at 643 cm–1 when irradiating monometallic Cu(I)–MCM-41 loaded with CO2. Furthermore, FT-Raman spectra of the ZrCu(I)–MCM-41 sieve did not show the characteristic absorption of clusters or particles of Cu2O (190, 154 cm–1), CuO (348, 299 cm–1), or ZrO2 (series of sharp bands in the 700–100 cm–1 region). Comparison of the noise level of the ZrCu(I)–MCM-41 Raman spectrum with the intensity of the 190 cm–1 peak of a mechanical mixture of Cu2O and MCM-41 allowed us to conclude that at most 10 percent, if any, of the Cu present in ZrCu(I) or Cu(I) sieve is engaged in Cu2O cluster formation.

(a) FT-IR spectrum of evacuated ZrCu(I)–MCM-41 (referenced against neat MCM-41). (b) Difference FT-IR spectra before and after photolysis (355 nm, 165 mW cm–2, 2.5 h) of CO2 loaded (1 atm) ZrCu(I)–MCM-41 (top), or exposure of the sieve to 1 atm O2 for 24 h in the dark (bottom trace).
The observed photoproducts CO and H2O (with the oxygen atom originating from CO2) imply that electron transfer from MMCT-excited Zr results in the dissociation of carbon dioxide. The transient O– so produced, in the presence of a high density of surface SiOH groups [24] will be protonated.
The surface OH radicals like the CO molecules will diffuse through the channels until they encounter an isolated Cu(I) site, resulting in reduction to H2O (diffusion of small species by random walk in nanoporous solids has recently been monitored in our laboratory by time-resolved step-scan FT-IR spectroscopy [25]). Therefore, we propose that CO2 splitting is the key mechanistic step at excited MMCT sites, consistent with the reaction path of single UV photon-induced CO2 reduction by H2O to CO and O2 in framework TiMCM-41 sieve [26]. This is the first observation of CO2 photoreduction at a binuclear MMCT site at the gas–solid interface.
4 Conclusions
In summary, assembly of Ti–O–Cu(I), Ti–O–Sn(II) and Zr–O–Cu(I) MMCT moieties on mesoporous silicate surfaces with each metal in a pre-selected oxidation state has been demonstrated. The method is based on the use of precursors with labile CH3CN ligands that are readily substituted by surface titanol or silanol groups under mild conditions. Optical and FT-IR spectroscopy allowed us to identify the Cu–O (Sn–O) bond modes associated with the visible Ti–O–Cu(I) and Ti–O–Sn(II) MMCT sites. Excitation of the MMCT chromophore of ZrCu(I)–MCM-41 loaded with 1 atm of CO2 gas at room temperature resulted in the production of CO and H2O, with the oxygen atom of water originating from CO2. This is the first observation of CO2 photoreduction at a binuclear MMCT site at the gas-solid interface. A Z-scheme for CO2 reduction by H2O under visible light can be envisioned if a metal M can be identified that oxidizes H2O by visible light-induced LMCT, thereby reducing the bimetallic moiety to its original state.
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical, Geological, and Biosciences of the US Department of Energy under contract No. DE-AC03-76SF00098.