

1 Introduction
In the last few years, phosphosilicate gels have attracted much attention and many works have been carried out. Indeed, these materials present interesting technological and structural properties suitable for fast proton conductor [1–3] and for applications in biomaterials and catalysts [4].
A route to synthesize this class of materials is the use of the sol–gel process allowing the preparation of inorganic oxides through hydrolysis and condensation of the precursors in solution at room temperature [5].
More recently, D’Apuzzo et al. [3] have reported the synthesis by sol–gel process of phosphosilicate gels having the molar composition 10 P2O5·90 SiO2 (10P) and 30 P2O5·70 SiO2 (30P) using POCl3 as phosphorous precursor and TEOS (Si(OCH2CH3)4) as silica source. They have studied their structures by a detailed solid-state 29Si, 31P and 1H SPE MAS NMR experiments for different heat treatments. It was found that the formation of Si–O–P bonds occurred for the (30P) gel and after heat treatment at 300 °C [6].
Moreover, the authors were interested in the spatial connectivities of 31P Qn resonances in the phosphosilicate gels. They established, by using two-dimensional NMR 31P magnetization exchange experiments, that the (10P) gel contained distinct domains with different phosphorous connectivities [7].
In this paper, we report the synthesis and characterization of phosphosilicate gels obtained by a sol–gel method using TEOS and H3PO4 as precursors. The aim of this paper is to study the influence of the heat treatments in the structure of the material and above all, to show evidence for the formation of Si–O–P bonds within the matrix through a precise investigation by solid state NMR.
The emphasis will be made on heteronuclear experiments involving 29Si and 31P nuclei. CP cross polarization process between 31P and 29Si nuclei will be clearly demonstrated, leading to the set up of 2D 31P–29Si CP experiments. Such experiments will be suitable for the direct evidence of spatial connectivities between nuclei.
2 Experimental
2.1 Sample preparation
The compounds were prepared by sol–gel process from tetraethoxysilane (TEOS, purchased from Fluka) and H3PO4 (orthophosphoric acid 85%, 33.0 mmol of H3PO4 purchased from Aldrich).
TEOS was diluted in ethanol, and then distilled water was added to the solution. The molar composition of TEOS/EtOH/H2O was 1:4:3 [8].
After a prehydrolyze stage (1 h) at room temperature, 1 equiv of H3PO4 was added to the mixture. The reaction is slightly exothermic.
The wet gels were obtained after 2 days at room temperature and after drying 2 weeks in air, the dry gels (pulverized into powders) were heated to 136 °C during 6 days (SiP-136) and to 100 °C for 30 h (SiP-100). High temperature phases heated at 500 °C (SiP-500) and at 800 °C (SiP-800) during 2 h were also investigated.
2.2 Characterization of phosphosilicate gels
XRD patterns were obtained with a Phillips diffractometer working with Cu Kα radiation: λ = 1.5418 Å, in step mode between 5° and 75° of 2 θ degrees and by using a step size of 0.04° and 4 s per step for the compounds heated at 136 °C.
Solid-state NMR experiments were carried out at B0 = 7 T, with ν0(31P) = 121.51 MHz and ν0(29Si) = 59.63 MHz (AVANCE 300 Bruker spectrometer). 7- and 4-mm Bruker probes were used for 29Si single pulse experiment (SPE) spectra of SiP-136 and SiP-100, respectively. A 4 mm triple resonance Bruker probe was used for 31P MAS, 1H SPE MAS, the different experiments of cross polarization and for the 2D heteronuclear correlation (HETCOR) experiment.
Samples were spun at the magic angle using ZrO2 rotors (5–15 kHz).
31P NMR chemical shift are referenced to 85% H3PO4 and to TMS for 29Si and 1H.
Among the different NMR techniques used to characterize the phosphosilicate gels, we have in particular:
- • the double cross polarization magic angle spinning (CP MAS) experiment, whose pulse sequence is shown in Fig. 1. As we can see, 1H irradiation was used for relaxation purposes because of its shorter T1 (when compared to 31P), followed by a double 1H→31P→29Si transfer using a ramp, namely, a variable amplitude cross polarization (VACP) [9] sequence during the contact time (tCP), in order to broaden the Hartmann–Hahn profile. Then, proton CW decoupling was applied during the 29Si acquisition;
- • the 2D CP MAS HETCOR experiment, whose pulse sequence is depicted in Fig. 2. This technique consists of an extension of the basic 1D CP.
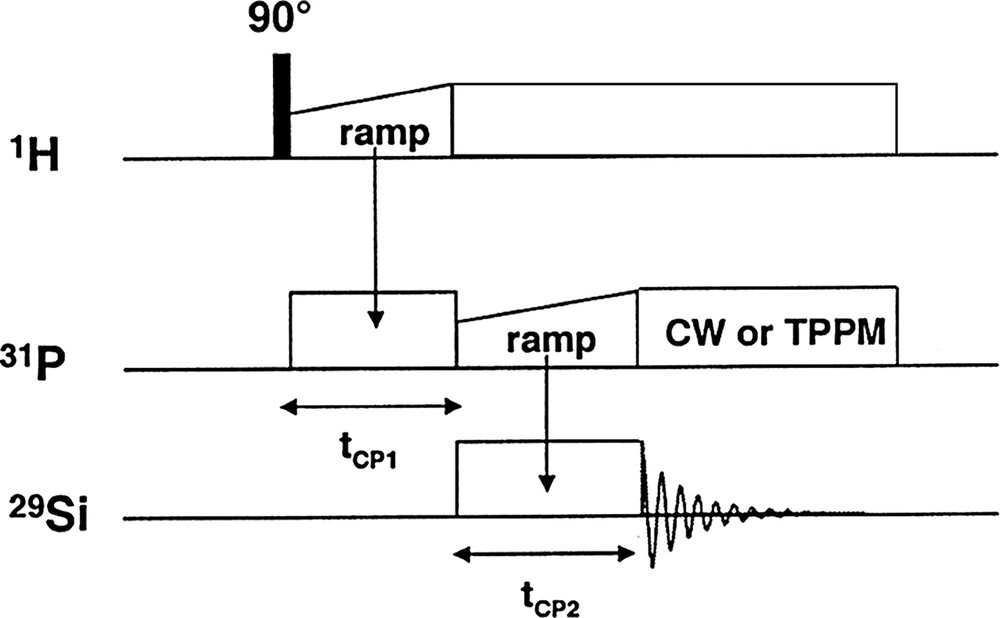
CP 1H→31P→29Si pulse sequence.
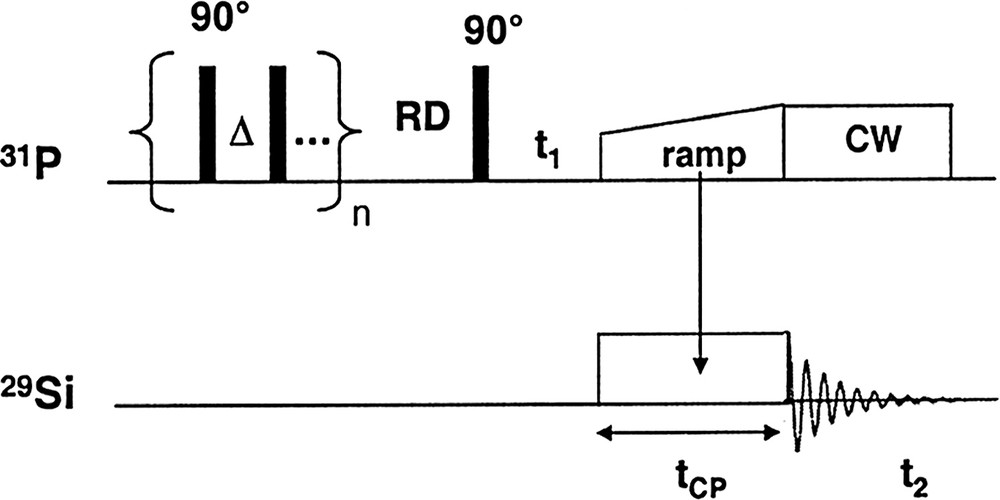
31P–29Si HETCOR pulse sequence.
Because of the long T1(31P) a presaturation on the 31P channel was applied, followed by a relaxation delay (RD) optimized to be the better compromise between the effective 31P magnetization and the spectrometer time. After the time evolution in t1, a cross polarization 31P→29Si was performed using a VACP sequence during the contact time. A CW 31P decoupling was used then during the 29Si acquisition.
3 Results and discussion
3.1 X-ray diffraction
Fig. 3A shows the XRD patterns of phosphosilicate gels heated at different temperatures, and Fig. 3B gives the attribution of the different peaks.
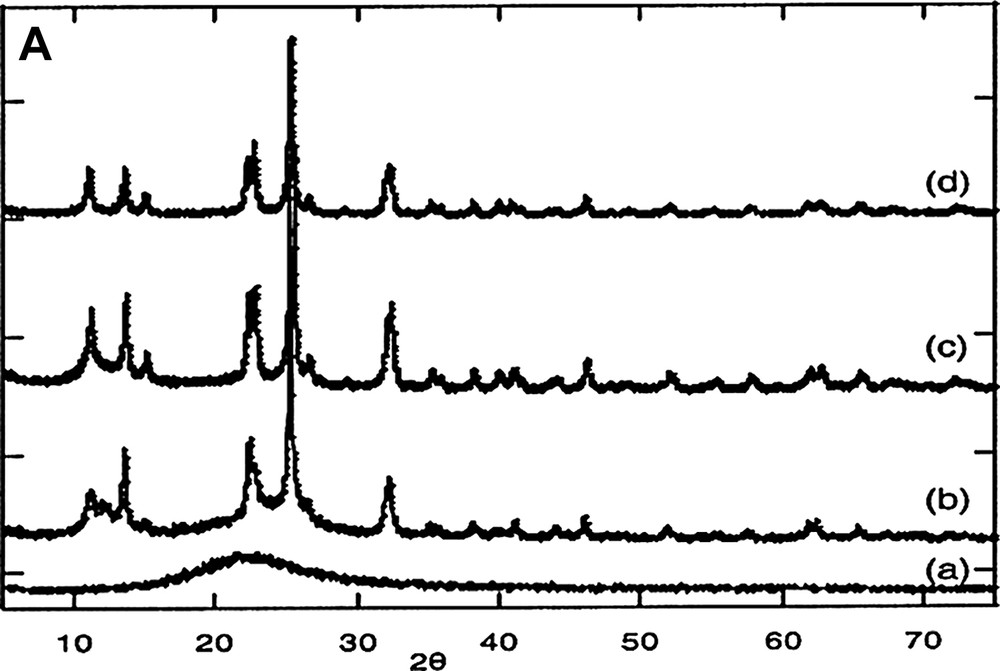
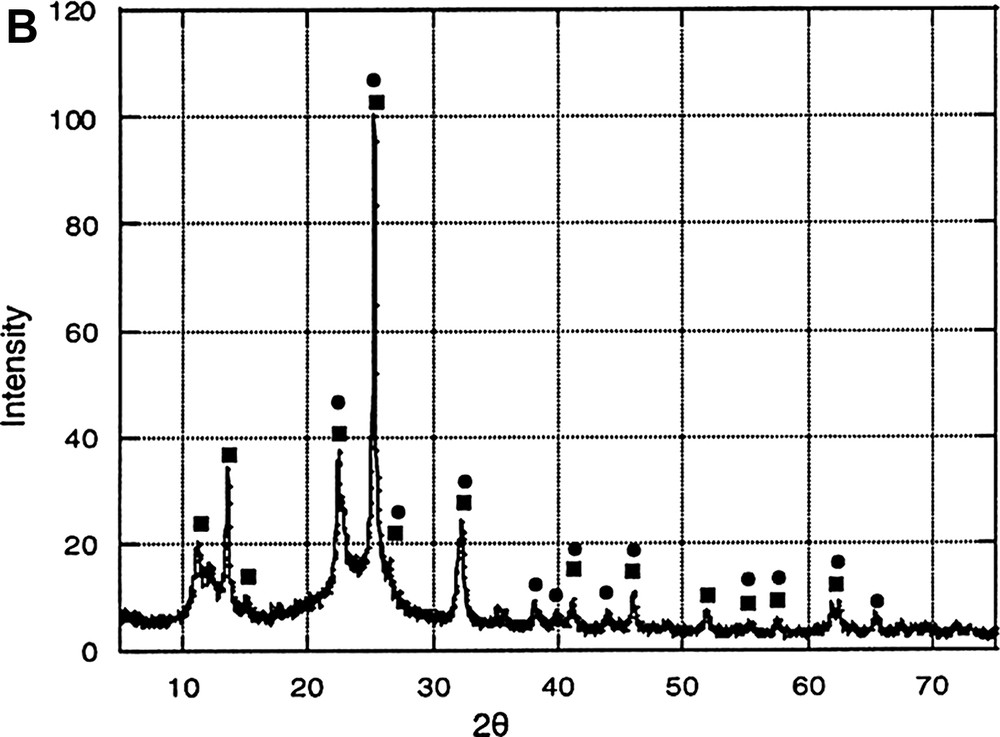
(A) XRD patterns of the gels heated at different temperatures: (a) SiP-100, (b) SiP-136, (c) SiP-500, (d) SiP-800. (B) XRD patterns of SiP-136: ■ Si5O(PO4)6 phase; ● Si(HPO4)2·H2O phase.
In the case of SiP-100, just a large peak corresponding to amorphous silica is observed.
However, after a heat treatment at 136 °C, several peaks appear clearly in the diagram, assigned to Si5O(PO4)6 [10] [11] and Si(HPO4)2·H2O phases [12].
At higher temperature, a unique phase (Si5O(PO4)6) is observed [11].
It seems from this X-ray powder diffraction study that the crystallization occurs between 100 and 136 °C. Such a low temperature was unexpected [6], showing the crucial role of the precursor used in the various synthesis.
All samples were further investigated by MAS, simple and double CP MAS experiments (29Si, 31P, 1H).
3.2 Magic-angle spinning nuclear magnetic resonance spectroscopy
3.2.1 1H MAS NMR
As we can see in Fig. 4, the 1H MAS NMR spectrum of the different gels are comparable. It shows a single resonance at 10.2 ppm assigned in majority to the POH groups, but we do not exclude the possible presence of SiOH groups and water molecules present notably in the Si(HPO4)2·H2O phase.
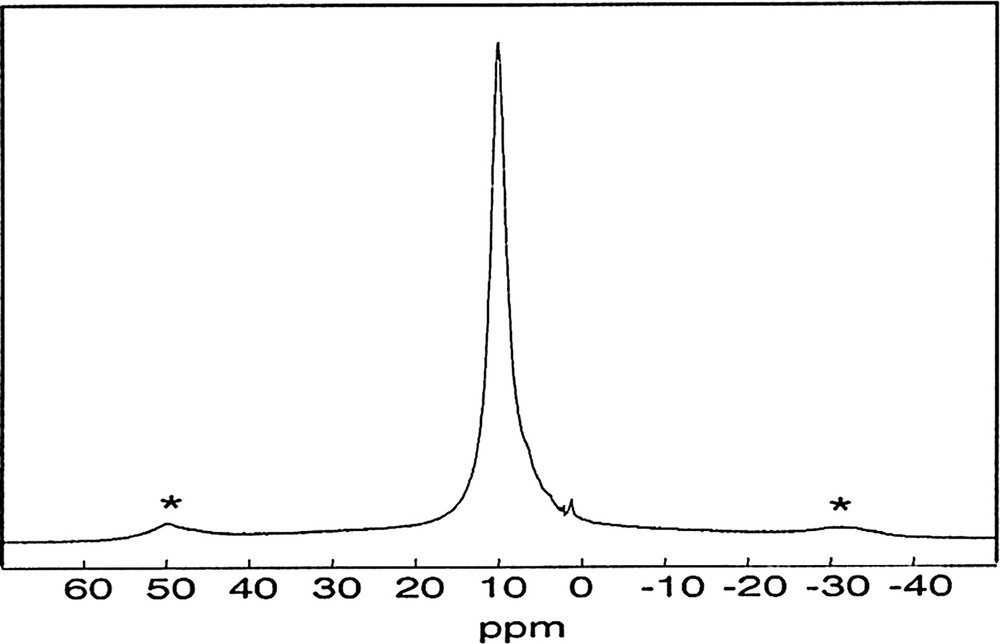
1H MAS NMR spectrum of SiP-136. (∅: 4 mm, RO: 12 kHz, NS: 4, RD = 10 s, 90° (1H): 2.5 μs).
Moreover, it is important to note the absence of resonance between 1 and 4 ppm, suggesting that the hydrolysis and the condensation of ethoxy groups from TEOS have occurred totally.
3.2.2 31P dec{1H} and 1H→31P CP MAS NMR
For SiP-100 (Fig. 5a, b), we can observe the presence of three resonances at 0 ppm, ~ –10 ppm and ~ –25 ppm attributed, respectively, to Q0 O=P(OH)3, Q1 O=P(OP/OSi)(OH)2 and Q2 O=P(OP/OSi)2(OH) units (Q1 and Q2 components are very minor. Their relative intensities are strongly increased by the CP process. Q0 units are underestimated under CP due to internal mobility) [13].
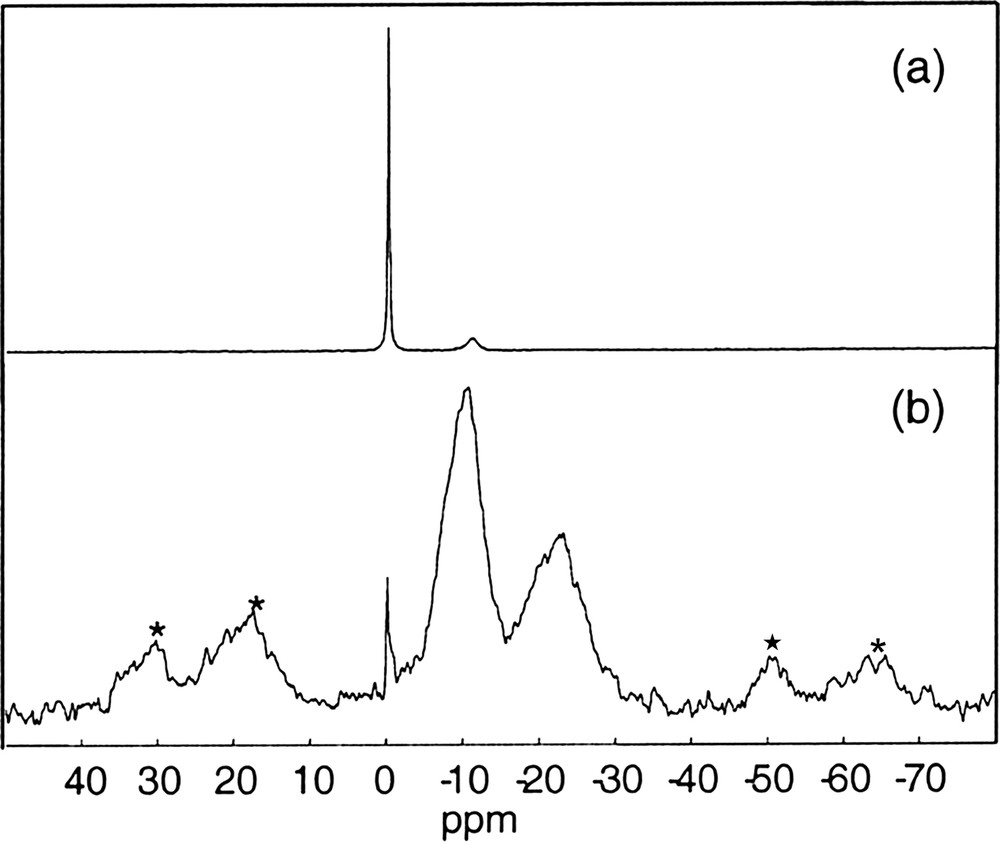
(a) 31P dec{1H} MAS NMR spectrum of SiP-100 (∅: 4 mm, RO: 14 kHz, NS: 204, RD = 10 s, 90° (31P): 9.0 μs, CW {1H} decoupling, LB = 10 Hz). (b) 1H→31P CP MAS NMR spectrum of SiP-100 (∅: 4 mm, RO: 5 kHz, NS: 616, RD = 10 s, 90° (1H): 2.6 μs, tCP = 2 ms, tppm {1H} decoupling, LB = 50 Hz). (c) 31P dec{1H} MAS NMR spectrum of SiP-136 (∅: 4 mm, RO: 12 kHz, NS: 44, RD = 10 s, 90° (31P): 9.0 μs, CW {1H} decoupling, LB = 30 Hz). (d) 1H→31P CP MAS NMR spectrum of SiP-136 (∅: 4 mm, RO: 5 kHz, NS: 40, RD = 10 s, 90° (1H): 3.7 μs, tCP = 3 ms, CW {1H} decoupling, LB = 30 Hz).
★ Spinning side bands.
Now, concerning the SPE 31P dec{1H} NMR spectra (Fig. 5c) of SiP-136, we can see the presence of several peaks. Besides the peak at 0 ppm corresponding to the orthophosphoric acid H3PO4 (Q0), additional resonances are present at –12.0, –30.9 and –35.0 ppm and assigned to Q1 O=P(OP/OSi)(OH)2 or Q2 O=P(OP/OSi)2(OH) and Q3 O=P(OP/OSi)3 units, respectively. Moreover, one resonance appears at –44.8 ppm assigned to Q4 units.
In order to obtain information about the protonation of these phosphorous units, cross polarization 1H→31P experiments have been performed as reported in Fig. 5d.
The spectrum shows clearly the disappearance of the peak at 0 ppm suggesting again that the Q0 species are strongly mobile in the silica matrix.
Concerning the resonance at –30.9 ppm, the intensity of lines permits to affirm that the associated Q3 units come from a protonated phase, such as Si(HPO4)2·H2O [14].
However, the chemical shift of the last resonance (–44.8 ppm), and the corresponding decrease of its intensity under CP experiment, are consistent with the isotropic chemical shift of the Si5O(PO4)6 phase [15].
At this stage of the analysis, it will be interesting to identify precisely the nature of the Q1 and Q2 units presents in the two samples (P–O–P and/or P–O–Si), by INADEQUATE 31P–31P experiments or 2D heteronuclear 29Si–31P experiments focused on these species.
3.2.3 29Si MAS, 1H→29Si CP MAS, 31P→29Si CP MAS, 1H→31P→29Si CP MAS NMR
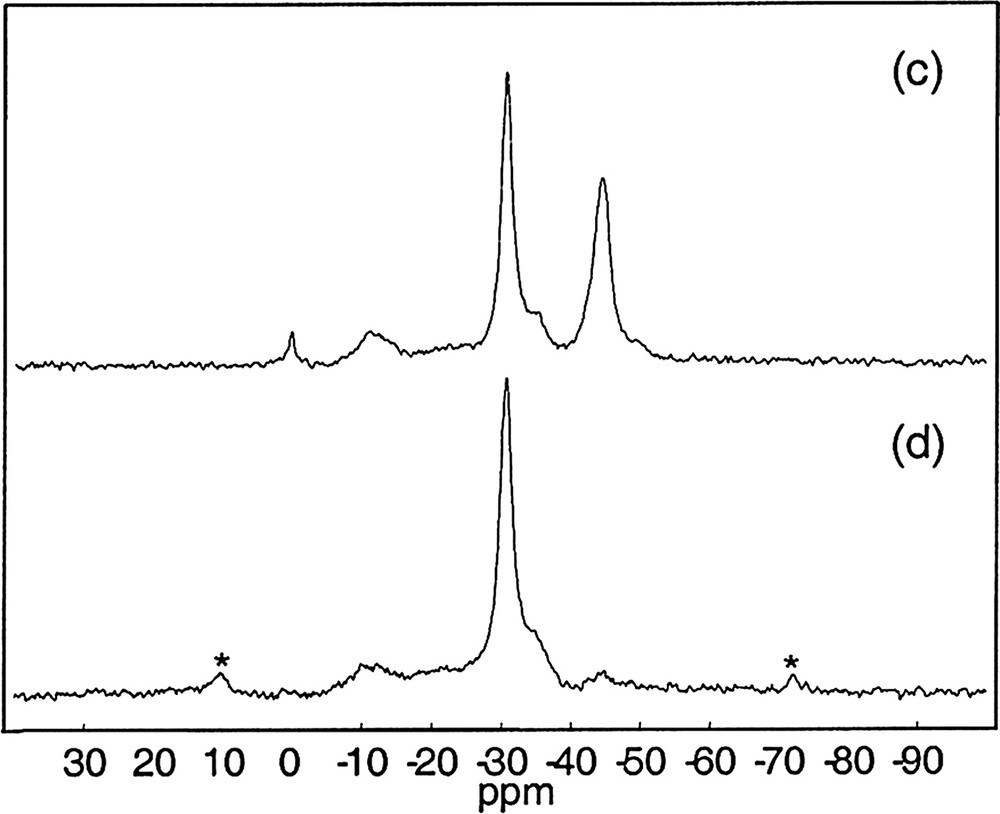
The Fig. 6 shows the 1H→ 29Si CP MAS NMR spectrum of SiP-100. This spectrum shows the presence of Q3 Si(OSi)3(OH) and Q4 (SiO4) units and is characteristic for condensed silica gels.
1H→29Si CP MAS NMR spectrum of SiP-100 (∅: 4 mm, RO: 5 kHz, NS: 5152, RD = 10 s, 90° (1H): 2.3 μs, tCP = 10 ms, tppm {1H} decoupling, LB = 100 Hz).
SiP-136 was investigated by 1H→29Si, 31P→29Si CP experiments, as well by 1H→31P→29Si double CP experiments (Fig. 7) (the double CP pulse experiment is derived from the sequence depicted in Fig. 1, {1H} decoupling was suppressed and {31P} decoupling was applied).
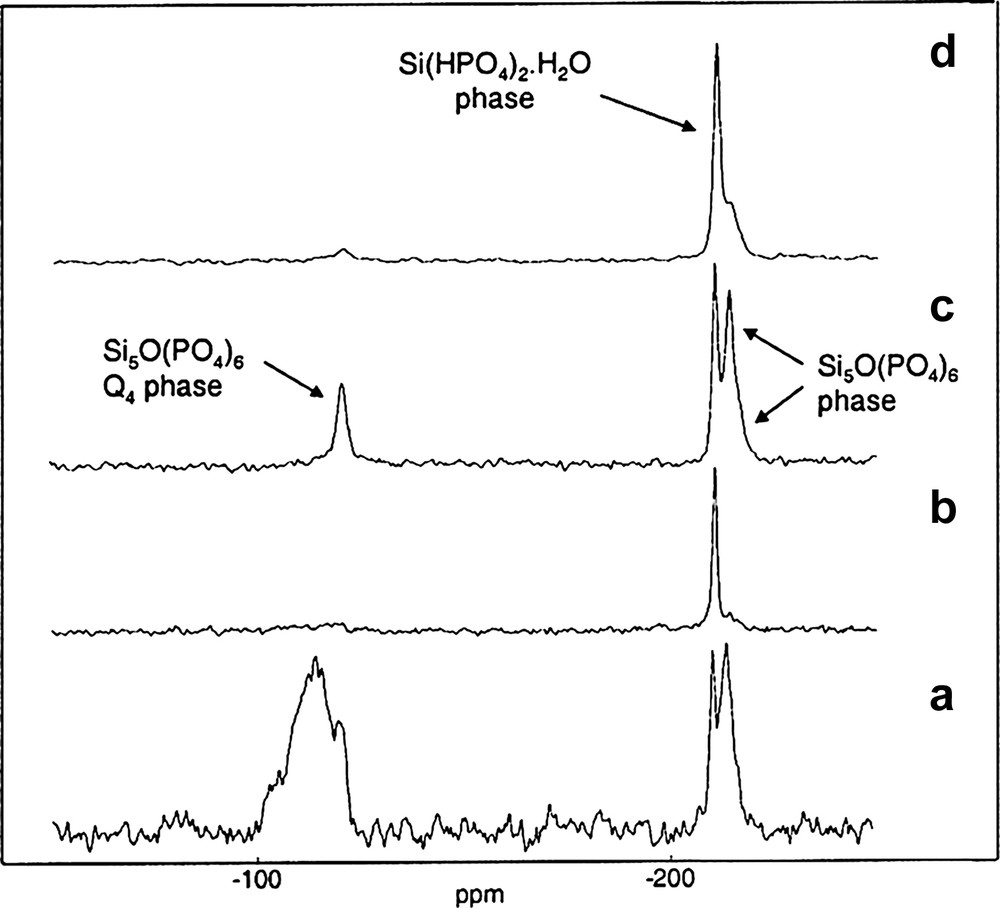
NMR spectra of SiP-136. (a) 29Si MAS (∅: 7 mm, RO: 4 kHz, NS: 481, RD = 120 s, 90° (29Si): 5.6 μs, LB = 41 Hz). (b) 1H→29Si CP MAS (∅: 4 mm, RO: 5 kHz, NS: 520, RD = 10 s, 90° (1H) : 5.4 μs, tCP = 40 ms, CW {1H} decoupling, LB = 30 Hz). (c) 31P→29Si CP MAS (∅: 4 mm, RO: 5 kHz, NS: 1680, RD = 30 s, 90° (31P): 5.8 μs, tCP = 40 ms, LB = 40 Hz). (d) 1H→31P→29Si CP MAS (∅: 4 mm, RO: 5 kHz, NS: 2880, RD = 10 s, 90° (1H): 3.7 μs, tCP1 = 3 ms, tCP2 = 40 ms, CW {31P} decoupling, LB = 60 Hz).
According to the different spectra shown in Fig. 7 and to the literature, the resonances at –120.5, –214.5 ppm and the shoulder at –217.0 ppm are assigned to the four and sixfold coordinated silicon atoms present in the Si5O(PO4)6 phase [15].
The narrow peak at –210.8 ppm is assigned to the sixfold coordinated silicons in the Si(HPO4)2·H2O phase [14].
Thanks to the 29Si SPE MAS spectrum (Fig. 7a), we can note also the presence of a broad resonance at ~ –110 ppm attributed to amorphous silica (Q4 units), not visible in the XRD patterns.
The 1H→29Si CP MAS experiment (Fig. 7b) is able to discriminate the two phases Si5O(PO4)6 and Si(HPO4)2·H2O by their protonation state. Besides, we have willfully used a long contact time (tCP = 40 ms), that is why the resonance corresponding to the amorphous silica was not detected.
The 31P→29Si CP MAS experiment (Fig. 7c) shows clearly the formation of Si–O–P bonds in the phosphosilicate gel SiP-136, by selecting the resonances of both crystalline phases Si5O(PO4)6 and Si(HPO4)2·H2O. In the Fig. 7d, the 1H→31P→29Si CP MAS experiment overestimates the Si(HPO4)2·H2O resonance as expected.
3.2.4 2D 31P–29Si HETCOR
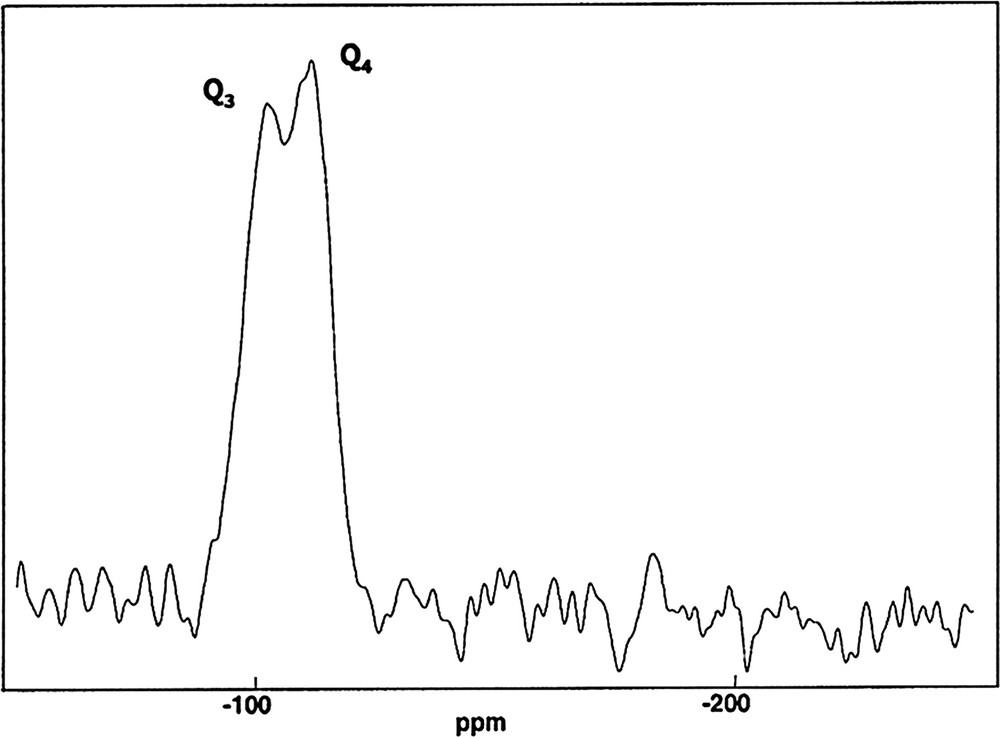
In order to deepen this study, we have performed a two-dimensional 31P–29Si HETCOR experiment, based on cross polarization, through dipolar coupling (as presented in Fig. 8). The pulse sequence is presented in Fig. 2.
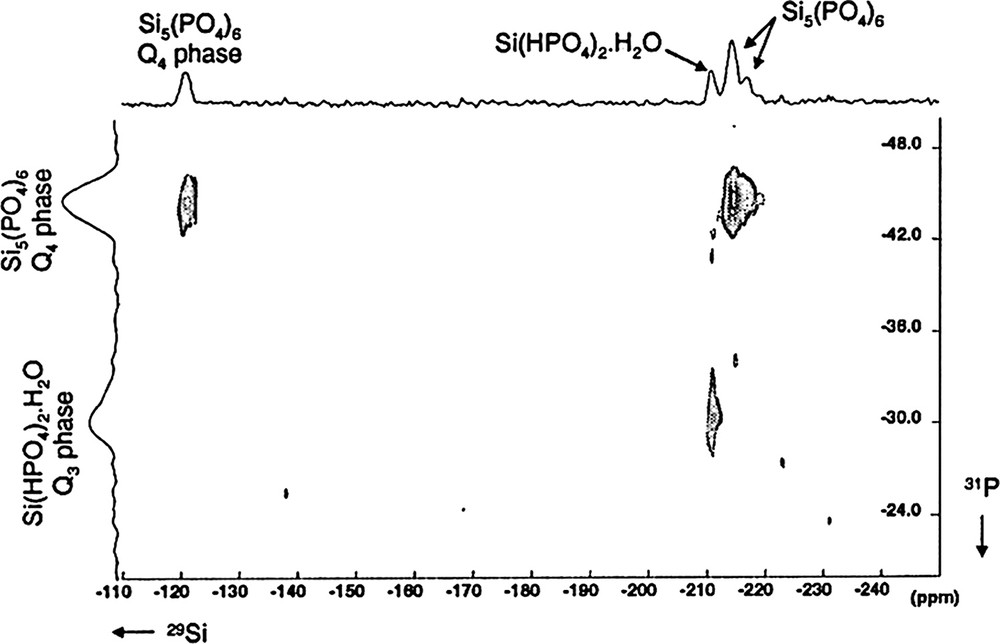
2D 31P→29Si (HETCOR) NMR map of SiP-136 (∅: 4 mm, RO: 5 kHz, NS: 64 for each t1 increment, Δ = 30 ms, n = 20, RD: 60 s, 90° (31P):5.8 μs, tCP = 40 ms, CW {31P} decoupling, 64 h, LB = 40 Hz in F2 and F1).
29Si resonances located at δ = –120.5, –214.5 and –217.0 ppm are clearly related to a unique 31P resonance located at δ = –44.8 ppm. The three correlation correspond indeed to the Si5(PO4)6 crystalline phase.
One more correlation is observed at δ(29Si) = –210.8 ppm and δ(31P) = –30.9 ppm: it is definitely assigned to the Si(HPO4)2·H2O phase.
These results show that 2D heteronuclear experiments can be safely used for the characterization of complex mixtures of phosphosilicates phases. It can be noticed that no correlation is observed at δ(31P) = –12.0 ppm not shown above. It tends to prove that this resonance can not be assigned to Si–O–P bonds.
However, the S/N ratio of the 2D experiment seems not sufficient for definite conclusions.
4 Conclusion
In this work, we have underlined the influence of the heat treatment in the structure of the silicophosphates gels. It was found by XRD and by a detailed analysis by solid-state 29Si and 31P NMR that the gel treated at 100 °C for 30 h had a structure consisting of a silica matrix including isolated molecules of phosphorous acid. The heat treatment at 136 °C permits the formation of Si–O–P bonds in the gel, evidenced by the 2D 31P–29Si CP MAS NMR experiments.
At present, it will be interesting both to go further into the characterization of the Si–O–P bonds, by doing a solid-state NMR analysis through the J-coupling, to prove unambiguously the chemical bonding between Si and P atoms in the gel state and to perform 31P–31P INADEQUATE experiments and 2D heteronuclear 29Si/31P experiments in order to identify the chemical nature of the Q1 and Q2 units. Works are in progress in both directions.