

1 Introduction
Isotope labelling is a powerful tool to elucidate metabolic pathways [1] and to determine metabolic fluxes [2]. Nuclear magnetic resonance (NMR) is widely employed to detect and quantify labelled compounds resulting from the metabolisation of a fed stable isotope labelled substrate. A major advantage of NMR compared to mass spectroscopy or radioactivity measurements is that crude cell extracts can be analysed without further sample purification or derivatisation, and that the detection is essentially indiscriminate with respect to chemical properties of the metabolites. Thus it is possible to detect all sufficiently abundant small soluble metabolites in their chemical diversity. The main drawback of NMR, however, is its rather low sensitivity restricting analysis to the more abundant metabolites, though this is sufficient for many purposes.
The most useful stable isotopes for NMR detected metabolic labelling experiments are 13C and 2H, elements which occur in nearly all metabolites, and 15N which is profitable for specifically following the metabolism of nitrogen containing compounds. Between 13C and 2H, the former is much more commonly employed. It has a wide chemical shift range spreading the signals out and reducing overlap between different peaks. Its sensitivity can often be improved by employing indirect detection schemes via attached protons. However, 13C has a natural abundance of 1.1%, which means that it can be difficult to distinguish small labelled metabolite pools from the natural abundance signals of major metabolites. On the contrary, 2H has a very low natural abundance, only 0.02%, so that even small signals from labelled compounds are easily picked out. However, when there are several labelled compounds present, peak overlap rapidly becomes a problem since the chemical shift range is narrow, and moreover, peaks are relatively large owing to the fact that 2H is a spin 1 nucleus with an albeit small quadrupole moment. Thus the strengths and weaknesses of 13C and 2H are quite complementary, and in certain cases it might be a good strategy to combine both labels.
In recent work [3] investigating the demethylation of nicotine in plants, we fed (R,S)-[13C-methyl]-nicotine to Nicotiana plumbaginifolia cells, and analysed extracts taken from cells after several days of culture with the labelled substrate. Several small peaks resulting from the metabolisation of the fed labelled nicotine could indeed be identified in 1D 13C and 2D 1H–13C HMQC spectra. Nevertheless this result was obtained only after careful comparison with spectra obtained from cell extracts after feeding of unlabelled nicotine, and by showing that the peaks of interest increased with the duration of the feeding experiment. Parallel feeding experiments with (R,S)-[2H3-methyl]-nicotine showed the unequivocal appearance of 2H-labelled metabolisation products but these could not be identified from the 2H NMR spectra. In order to push the investigation further we are now conducting feeding experiments with doubly labelled (R,S)-[13C2H3-methyl]-nicotine instead of using the isotopes separately. Of particular interest will be the question of whether the methyl-group is transferred as a whole, leading to 13C2H3-containing compounds, or only after oxidation, leading to 13C2H21H- or13C2H1H2-containing compounds.
The feasibility of a NMR detected 13C2H3 double labelling strategy for plant metabolic studies has recently been shown in the clarification of the biosynthesis pathway for artemisinin in Artemisia annua [4]. The NMR method chosen for this work [5] was 13C-detected heteronuclear 13C–2H 2D correlation spectroscopy (COSY, sometimes also termed HETCORR). However, this method does not produce pure absorption peaks and has to be treated in absolute mode. A gain in sensitivity might therefore be expected from other methods, and the aim of the present paper is to explore different possible pulse sequences in order to choose the most sensitive for the planned metabolic study.
2 Material and hardware considerations
(R,S)-[13C2H3-methyl]-nicotine (see below) was synthesised from nornicotine and [13C2H3]-methyliodide as described by Mesnard et al. [3]. 6 μl of labelled nicotine were dissolved in 500 μl of H2O and used directly for the NMR measurements.
NMR analysis was carried out on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 125.69 MHz and 76.77 MHz for 1H, 13C and 2H respectively. Two probeheads were used with similar results, a 5-mm broadband inverse detection (BBI) probehead and a 5-mm triple-resonance 13C–15N (TXI) probehead. In both cases, the lock circuit was used for 2H and the broadband or 13C circuit for 13C. Spectra were run in unlocked mode, since the spectrometer was not equipped for using another lock nucleus than 2H. 90° pulse lengths for 13C were 14.6 μs (powerlevel –3 dB) with the BBI probehead and 10.8 μs (powerlevel –5 dB) with the TXI probehead. The 2H circuit was quite prone to arcing on both probeheads and powerlevels of +3 dB had to be used leading to 90° pulse lengths of 25.1 and 23.0 μs on the BBI and TXI probehead, respectively. Trials to tune and match the 2H circuit, which can be done only when the probehead is taken out of the magnet did not lead to any great improvements.
The broad-band preamplifier was used for both 13C and 2H detection. The 2H preamplifier had to be physically disconnected to avoid a leakage lock signal in the 2H spectrum (even when the lock mechanism was switched off by software control).
For experiments using both nuclei 13C and 2H, the spectrometer had to be equipped with a third radiofrequency channel, since on this Bruker spectrometer series, either channel 1 or channel 2 has to be on 1H. Both 13C and 2H channels also had to be equipped with filters to avoid spectral noise at the detection frequency coming from the decoupling channel. The 13C line was equipped with a standard 2H-stop 13C-pass filter and also a standard 1H-stop. Not disposing of a standard filter for the 2H line, we used instead a 13C-pass 31P-stop filter from a 300 MHz spectrometer. The frequencies were sufficiently close (75.47 vs. 76.77 MHz and 121.49 vs. 125.69 MHz, respectively) to those required that the 2H pulse length was not affected by more than 0.4 μs and that the noise introduced at the 13C frequency from 2H-decoupling completely vanished. Without the filter, the decoupling noise was about 150 times stronger than the normal noise level. The 1H line contained the usual 1H high pass but this was not strictly necessary.
3 Spectroscopy results and discussion
The one-pulse undecoupled 13C spectrum (Fig. 1) of the doubly labelled nicotine showed the expected 1-3-6-7-6-3-1 septuplet due to scalar coupling with 1JC–2H = 20.3 Hz. With 2H-decoupling and the proper frequency filters in the rf lines the septuplet collapsed and the 13C peak narrowed showing a pseudo-triplet of 4.3 Hz due to two distinct 3JC–H couplings of respectively 4.2 and 4.4 Hz (as taken from 1H spectra (data not shown)). When 1H-decoupling was applied simultaneously, the pseudo-triplet collapsed into a narrow singlet line.

Undecoupled (bottom) and 2H-decoupled (top) 13C-spectra of 0.6% v/v (R,S)-[13C2H3-methyl]-nicotine in H2O. NMR parameters: 90° pulse, 2.7 s interpulse delay, 64 scans, no 1H-decoupling, WALTZ-16 2H-decoupling where appropriate.
Similarly, the 2H spectrum in Fig. 2 shows the expected doublet separated by 1JC–2H and decoupling collapsed it as well. Compared to the 13C spectrum, the 2H spectrum had a roughly 10% better signal-to-noise ratio when taking into account the different number of scans. Indeed, the 2H equilibrium magnetisation M0 is expected to be higher than the 13C one due to the fact that 2H is a spin I = 1 nucleus which more than compensates for the lower gyromagnetic ratio γ. In fact, M0 is proportional to γ I (I + 1), giving a ratio M0(2H)/M0(13C) = 1.63, and furthermore the methyl group contains one 13C but three 2H. However, detection is for a given quality factor 2.37 times more sensitive for 13C than for 2H as it is proportional to γ7/4. Finally the quality factor of the resonance circuits depends on the fill factor, which is better for the inner coil (in favour of 2H in our case as the probeheads are for inverse detection with 13C on the outer coil and 1H and 2H on the double tuned inner coil) and on the quality of the resonance circuit [6]. The latter is usually good for the 13C circuit but not necessarily for the 2H circuit since mostly the quality of the 1H circuit is optimised at the expense of the 2H lock circuit.
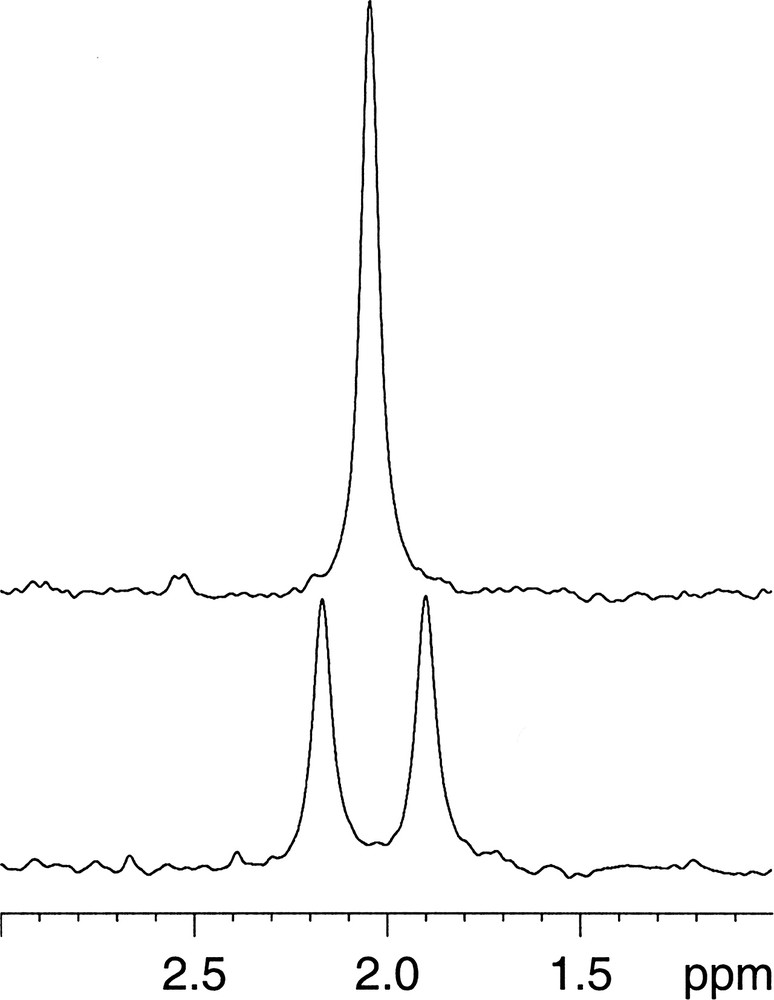
Undecoupled (bottom) and 13C-decoupled (top) 2H-spectra of 0.6% v/v (R,S)-[13C2H3-methyl]-nicotine in H2O. NMR parameters: 90° pulse, one scan, 13C decoupling with the GARP sequence.
The relaxation times for both nuclei were roughly evaluated. For 2H, a T1 of less than 0.2 s was found, whereas for 13C, T1 was around 3 s. This fast relaxation adds to the higher equilibrium magnetisation for 2H, and it can be argued that an optimal pulse sequence should start with the higher 2H magnetisation after only a short relaxation delay, and then transfer the polarisation to 13C for more sensitive detection, preferably with a direct detection probehead which however we do not have. It might seem tempting to also make use of the 3JC–H couplings but besides their relatively small value not allowing for an efficient transfer, this coupling is specific for nicotine, and is not general for other molecules that take over the methyl group : for example, methionine, which was shown to incorporate the methyl group from nicotine [3], does not have such a coupling.
Given the previous arguments for a transfer from 2H to 13C which was already chosen for the artemisin in work [5] and the advantage of having pure absorption peaks, the nearest possibility was to expand the heteronuclear COSY sequence by adding 180° refocusing pulses in the middle of the delays thus getting the INEPT sequence [7] (Fig. 3). This sequence has indeed already been used earlier in its 1D version for investigating different aspects of polymerisation in plastics after 2H labelling and polarisation transfer onto natural abundance 13C [8] and then in its 2D version for assigning 2H resonances of lowly 2H-enriched molecules in solution [9]. More recently, the technique has found quite widespread use for assigning and measuring quadrupolar couplings for 2H in oriented media using either cross polarisation [10], HMQC-type experiments [11] or INEPT and DEPT transfers [12]. Also preliminary work on the metabolisation of the herbicide glyphosate by soil micro-organisms has been presented [13] but this does not seem to have been followed up.
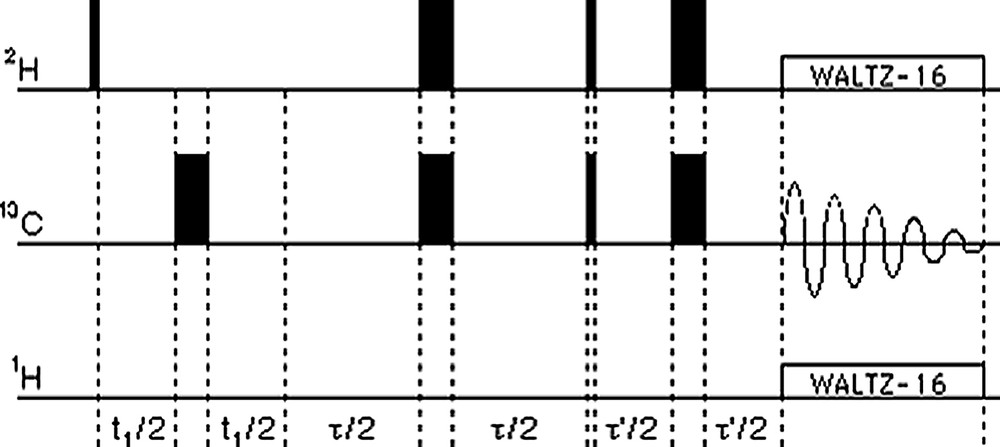
The 1H-decoupled 2H–13C 2D INEPT pulse sequence. Quadrature detection in the f1 dimension uses the TPPI scheme (π/2 increment of the phase of the second 2H 90° pulse). The 1D version is obtained by omitting the t1 block.
We first optimised the transfer τ and refocusing τ′ delays used in the INEPT sequence. For the transfer delay τ during which 213Cz2Hx evolves, the theoretical value of 1/(2 J) = 24.6 ms for the whole delay (i.e. before and after the 180° pulse) worked best which means that relaxation losses were negligible. According to Brown [5] the optimal refocusing delay τ′ for the COSY sequence was close to 1/(7 J), and we found indeed the optimum between 5.5 and 7 ms, i.e. between 1/(9 J) and 1/(7 J). The optimised 1D INEPT sequence was then 50% more sensitive than the one-pulse 13C spectroscopy as can be seen in Fig. 4. The small natural abundance peaks from the pyrrolidine ring that are visible in the one-pulse spectrum are effectively suppressed as are those from the aromatic pyridine ring.
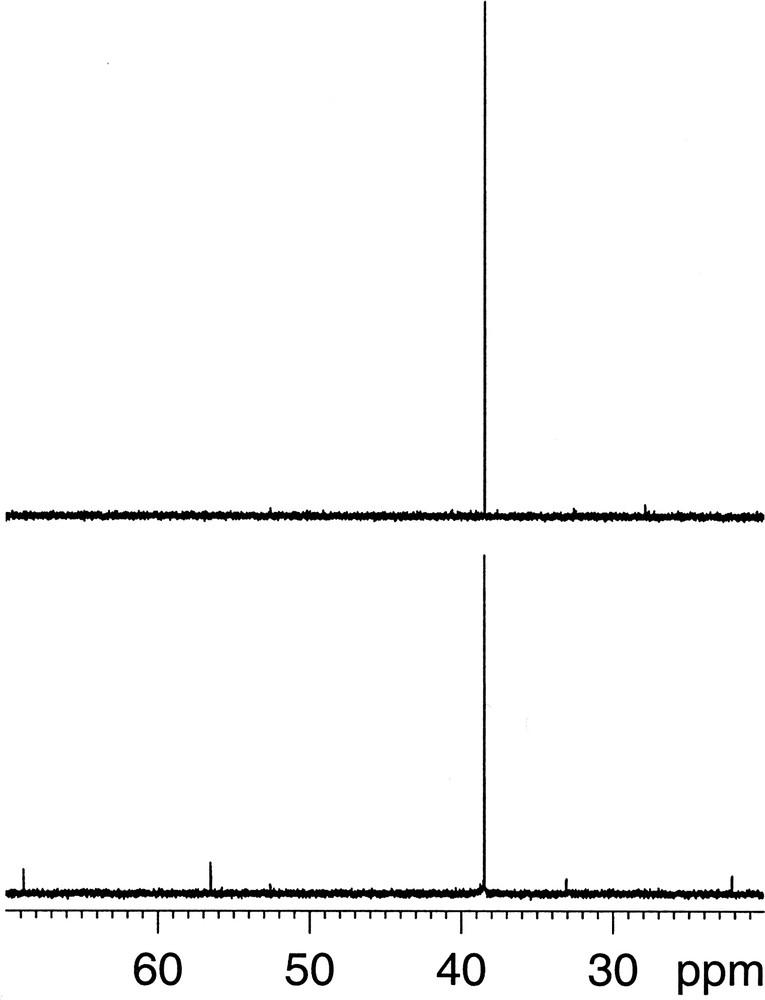
Single-pulse 13C (bottom) and 1D 2H–13C INEPT (top) spectra of 0.6% v/v (R,S)-[13C2H3-methyl]-nicotine in H2O. NMR parameters: 90° pulse, 1.7 s interpulse delay, 16 scans, WALTZ-16 2H- and 1H-decoupling; specifically for the INEPT: 24.6 ms transfer delay τ, 7.0 ms refocusing delay τ′.
The spectrum resulting from the corresponding 2D INEPT sequence can be seen in Fig. 5. Some t1-noise is apparent, probably due to the fact that the spectrometer was run in unlocked mode. Its magnitude can be appreciated from the projections, which show the 1D spectra corresponding to the frequency of the peak maximum. It should be noticed that sufficient resolution was achieved with relatively few increments since the 2H chemical shift range is small. When 13C is the indirect dimension, good resolution takes many more increments thus increasing problems with t1-noise. We also tried HSQC spectra in both directions (13C → 2H → 13C or 2H → 13C → 2H) and heteronuclear COSY sequences but never got as good results as with the INEPT sequence (data not shown). This is not surprising as shown by the sensitivity considerations above and by previous workers with other systems [8,9,12]. 13C → 2H → 13C HSQC spectra can’t be more sensitive than one-pulse 13C spectra as the excited and detected nucleus are the same, and there will rather be transfer losses so the demonstration of Fig. 4 is also valid for this case. The opposite transfer 2H → 13C → 2H suffers from the lower sensitivity for detection compared to 13C but depending on the probehead this effect is not very strong. The advantage of HSQC-type transfers is that their efficiency does not depend on the number of 2H attached to the 13C so that quantification of relative populations of 13C2H3, 13C2H21H and 13C2H1H2 isotopomers is more reliable. Identification of these groups should be possible just from the 2H induced isotope shift of the 13C resonance but if this is not sufficient then DEPT transfers might also be of interest.

2D 2H–13C INEPT spectrum of 0.6% v/v (R,S)-[13C2H3-methyl]-nicotine in H2O. The full spectrum is shown, and the projections correspond to the respective 1D spectra at the frequency of the nicotine methyl cross peak. NMR parameters: 0.1 s relaxation delay, 24.6 ms transfer delay τ, 7.0 ms refocusing delay τ′, four scans per increment, 256 increments for 10 ppm spectral width giving an FID resolution of 3.0 Hz, TPPI quadrature, WALTZ-16 1H- and 2H-decoupling during acquisition, 8 min total duration.
The 2D 2H–13C INEPT sequence, if necessary completed by other transfer sequences, thus seems a promising tool to analyse the metabolic fate of the doubly labelled methyl group from nicotine in plant cell suspensions and hopefully to learn more about the involved reaction mechanisms.
Acknowledgements
TAB wishes to thank the GERM committee for a grant towards her participation at the 19th annual GERM meeting, as well as the ‘Ministère des Affaires étrangères (France)’ for a studentship.