

The three-dimensional structure of [60]fullerene and the 30 reactive double bonds of its carbon network have inspired chemists to design and synthesize novel building blocks with unique chemical and physical properties. The spherical, all-carbon soccer ball has attracted an enormous research activity since the birth of the third allotropic form of carbon with hundreds of literature reports dedicated to the derivatization of the fullerene C60 [1]. A widely used method for the functionalization of C60 is the nucleophilic cyclopropanation of the [6,6] double bonds via the Bingel [2] reaction. For a single attack at a [6,6] bond one regioisomer is possible as the 30 double bond of the fullerene cage are equivalent. For a second attack, eight regioisomeric adducts (Fig. 1) are possible whereas, for trisadditions the number of the possible regioisomers increases to 46. The regioselective synthesis, isolation, and characterization of the different regioisomeric fullerene bisadducts and a limited number of trisadducts were firstly achieved by the stepwise cyclopropanation of C60 with diethyl bromomalonate [3]. The multiple and tedious chromatographic separation and purification steps by means of preparative HPLC required a more sophisticated approach to access polyadducts of C60 with well-defined three-dimensional structure. The tether-directed remote functionalization introduced by Icaacs et al. [4] was proved to be the method of choice and initiated a huge synthetic effort targeting a diversity of structurally different tethers in the first step and secondly, the facile access to fullerene addition patterns that had never been reported before. According to this concept (Fig. 1), the reactive malonate groups are connected with a more or less rigid organic moiety via spacers and thus, the tether directs the reactive groups to specific positions. The possibility of tuning the rigidity of the tether core as well as the spacer’s length has enriched the literature with a plethora of tethered systems that showed in many cases excellent regioselectivity in the Bingel functionalization of C60. However it should be mentioned that only opened-structure tethers were utilized for the regioselective remote functionalization of the C60 where, the malnate reactive groups are ‘hanged’ at the edges of the tethers. Furthermore, tethers designed for the synthesis of trisadducts of C60 required multiple synthetic steps or led to low yield functionalization reactions [5].
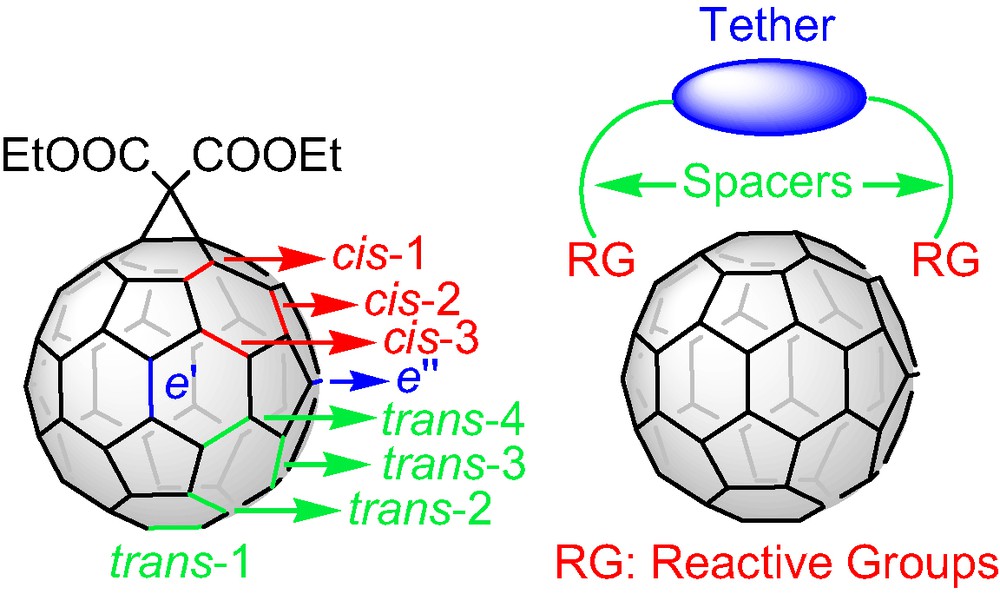
Position notation for bisadducts of C60 and the ‘tether-directed remote functionalization’ concept.
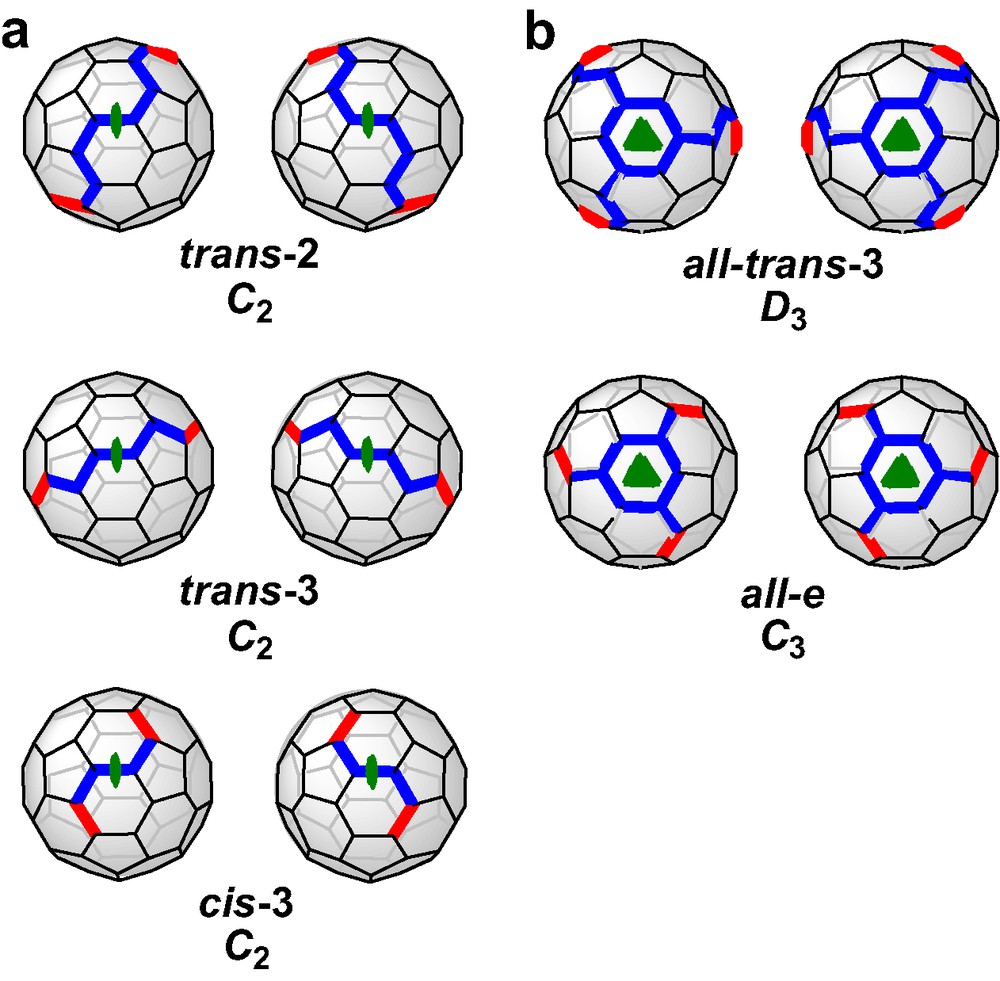
Recently, we reported [6] a new concept of the tether approach which was proved to be advantageous from different points of view. The rigid spacers were replaced by flexible alkyl chains and the malonate groups were incorporated in a macrocyclic ring. This way, the regioselectivity is not based on steric preorganization, but on the avoidance of unequal strain in the alkyl chains. The consequence for the symmetry of the addition pattern of the fullerene adducts is that macrocycles with two or three malonate units are forced to form adducts with rotational symmetry, that is C2 for bisadducts and C3 for trisadducts. All other regioisomers are excluded since they would result in different distances between the ester groups and thus, introduce strain. Fig. 2 shows the addition patterns of bis- and trisadducts that fulfill these restrictions. It becomes obvious that the regioselectivity of the Bingel functionalization can be adjusted by varying the length of the alkyl chains, a fact that accounts for the high versatility of this method. The concept of the cyclo alkylmalonates is not limited only for the synthesis of fullerene multiadducts containing rotational axes. If mixed macrocycles with two alkyl spacers of different length are used, bisadducts with Cs-symmetrical addition pattern are expected to form as outlined in Fig. 2.
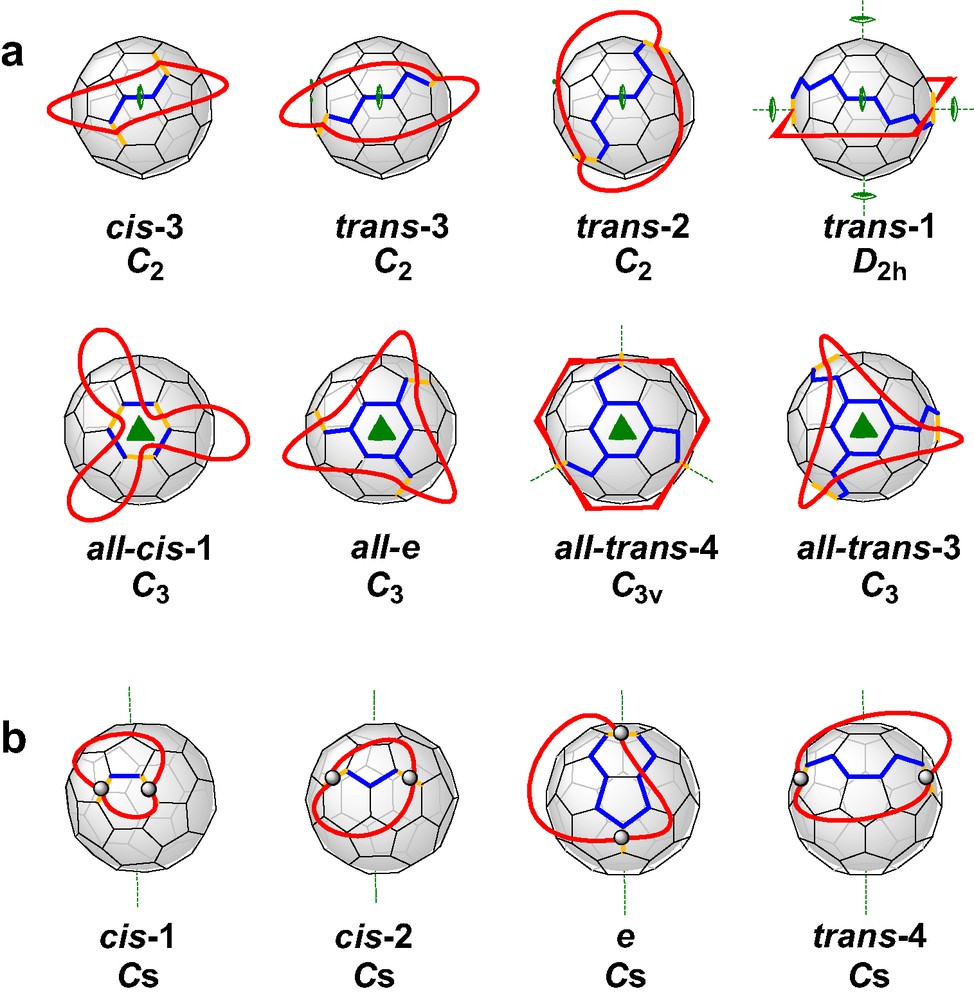
(a) Possible bis- and trisadducts of C60 containing rotational axes. All distances between adjacent addend positions are identical. (b) Bisadducts of C60 with Cs symmetry. The possible distances between adjacent addend positions are different. The yellow bonds denote the positions of addends, such as malonates.
The first advantage of the new concept presented was the synthesis of the proposed macrocyclic malonates which was proved to be quite simple. The desired cyclo alkylmalonates were obtained by the condensation reaction of malonyl dichloride with the appropriate diol in CH2Cl2 solvent and using pyridine as a base (Scheme 1). The reaction was performed under high dilution conditions to give a facile access to a great number of cyclo-[n]-alkylmalonates varying in the length of the alkyl chains and the ring size. The mixed macrocycles with two alkyl spacers of different length were prepared by the same method, using two different diols in the condensation reaction. The purification of the macrocycles could be accomplished by flash column chromatography on silica gel and the newly synthesized cyclic compounds were studied for the regioselective remote functionalization of [60]fullerene.
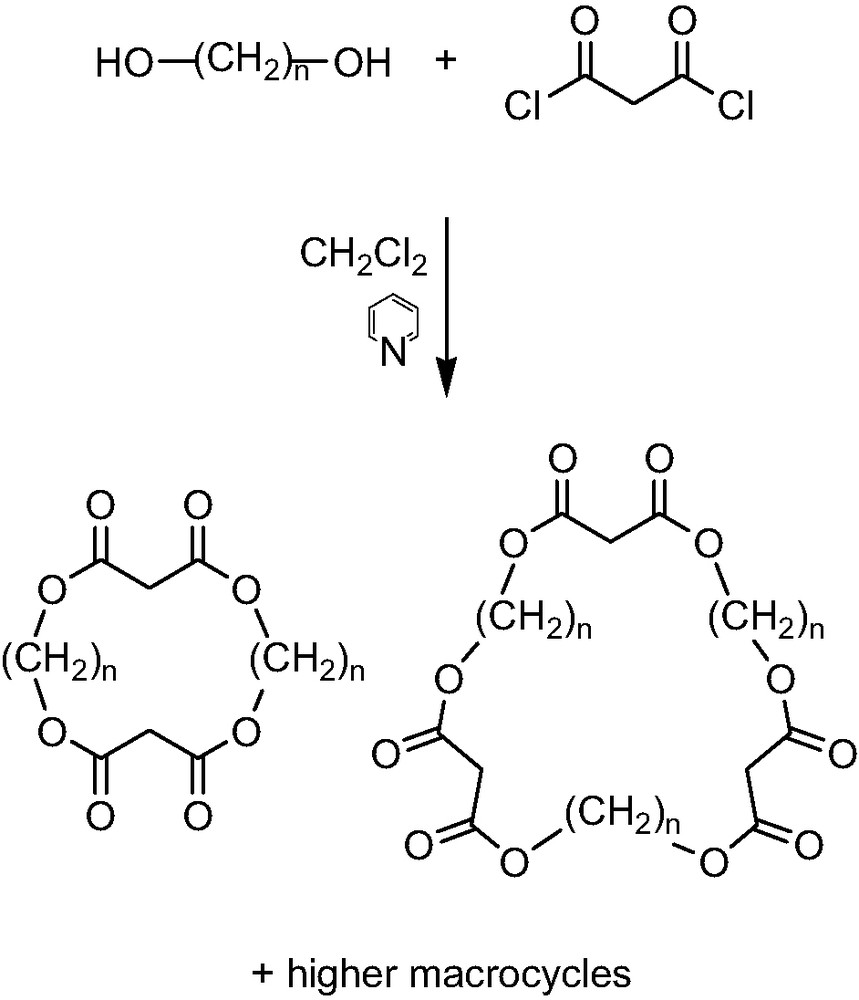
Synthesis of the cyclo-[n]-alkylmalonates.
The Bingel functionalization of C60 with the bis-malonate tether bearing C8 alkyl spacers was unsuccessful leading to the formation of polymeric material. Obviously, this macrocycle is not suitable for the formation of strain-free bisadduct with rotational symmetry because of the short length of the spacer units. The bis-malonate macrocycles with C12 and C16 alkyl spacers were treated with C60 in separate experiments under the Bingel conditions and the formed fullerene bisadducts were separated and characterized. The cyclo-[2]-dodecylmalonate afforded cleanly bisadduct trans-3 in a completely regioselective manner and in a remarkable 56% isolated yield (Scheme 2). Similarly, the cyclo-[2]-hexadecylmalonate bearing longer alkyl spacers reacted successfully with C60 leading to the formation of the trans-3 and trans-1 bisadducts, in a 55:45 ratio. The two regioisomers were isolated by flash column chromatography on SiO2 in 12.9% and 9.1% yields, respectively (Scheme 2). These results confirmed that the remote functionalization of C60 utilizing cyclic malonates proceeds with high degree of selectivity and more interestingly, this approach allows the access of different addition patterns by simple variation of the alkyl spacers.
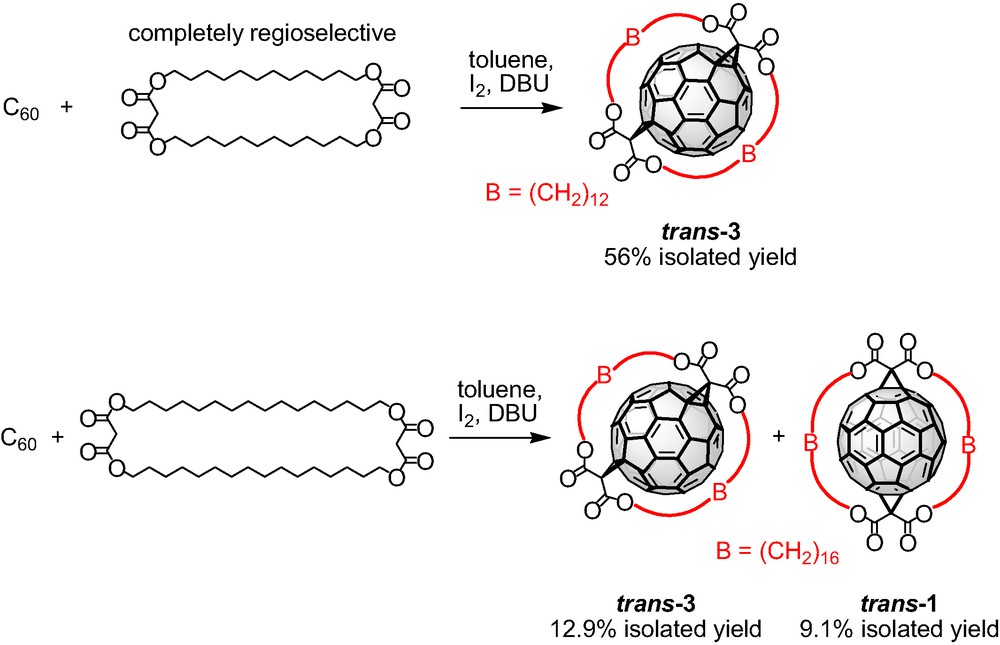
Tether-directed remote functionalization of C60 with the bis-malonates bearing C12 and C16 alkyl spacers.
Targeting the regioselective synthesis of Cs-symmetrical bisadducts of C60 we investigated the Bingel functionalization of the mixed bis-malonates that bear two alkyl spacers of different length. The results are demonstrated in Scheme 3. Both macrocycles gave a complete regioselective reaction with C60 and high yields of the formed bisadducts. Specifically, the bis-malonate tether bearing C4 and C8 alkyl spacers favored exclusively the cis-2 addition pattern while, the mixed macrocycle with C8 and C14 alkyl spacers showed complete regioselectivity for the equatorial fullerene bisadduct. In both cases the isolated yields were remarkably high.
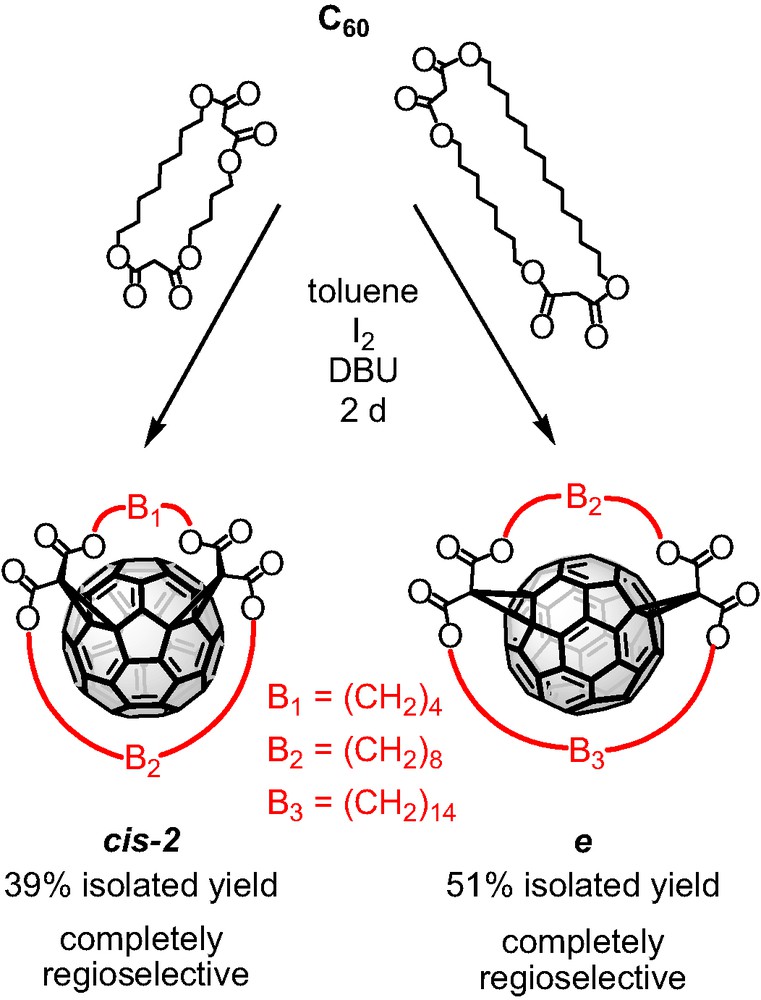
Tether-directed remote functionalization of C60 with the bis-malonates bearing C4–C8 and C8–C14 alkyl spacers.
As the regiochemistry of trisadditions on C60 is more complicated because of the large number of the possible regioisomers we applied the concept of the cyclo-malonates targeting trisadducts with well-defined structure. It was mentioned before that the symmetry of the tris-malonates synthesized is expected to favor the formation of trisadducts of C60 possessing threefold rotational symmetry, namely cis-1,cis-1,cis-1, e,e,e, trans-3,trans-3,trans-3 and trans-4,trans-4,trans-4. The formation of cis-1,cis-1,cis-1 is expected to be energetically disfavored because of the steric crowdness of the malonate addends. In order to confirm our theoretical predictions we firstly investigated the Bingel functionalization of C60 with the trimeric macrocycle bearing C8 alkyl spacers (Scheme 4). The reaction afforded the e,e,e trisadduct as the major product in 96% relative yield and the trans-4,trans-4,trans-4 as the minor in 4%. The separation of the adducts was accomplished by preparative HPLC and the two regioisomers were isolated in 40.2% and 2% yields, respectively. It should be highlighted that the trans-4,trans-4,trans-4 addition pattern was synthesized and characterized for the first time. Obviously, the C8-trismalonate tether fulfilled the expectation for the regioselective synthesis of trisadducts and prompted us to target a more remote addition pattern than the e,e,e. Thus, we subjected the macrocyclic tris-malonate bearing C14 alkyl spacers in a threefold Bingel cyclopropanation with the fullerene carbon sphere. The remote functionalization functioned in an excellent way and delivered the trans-3,trans-3,trans-3 regioisomer in a complete regioselective manner (Scheme 4).

Tether-directed remote functionalization of C60 with the tris-malonates bearing C8 and C14 alkyl spacers.
The concept of the macrocyclic malonates in the tether-directed remote functionalization of C60 was proved to be a powerful method to access fullerene polyadducts in a regioselective manner and in very satisfying isolated yields. The facile synthesis of the poly-malonates, their ability to show excellent regioselectivity, and the control of the regioselectivity by tuning the alkyl spacer’s length offered a new and effective method in this area of fullerene chemistry. In 2003, Zhou et al. [7] modified this approach by replacing the alkyl chains with polyether linkers and by using 3He@C60 instead of pristine C60. In an elegant way, the specific formation of C60 multiple adducts was monitored by 3He NMR. The bis-malonate macrocycle with tri-ethylene glycol spacers (Scheme 5) afforded regioselectively the cis-3 bis-adduct of C60 in 60% yield. 3He NMR analysis of the crude reaction mixture confirmed the formation of a single bis-adduct showing only one sharp peak at –10.236 ppm which is the precise chemical shift for the cis-3 fullerene bis-adduct of diethyl malonate. Similarly, the tris-malonate tether reacted with C60 to afford only one trisadduct isomer as confirmed by the 3He NMR spectra that showed one sharp peak at –12.040 ppm (Scheme 5). Its addition pattern was assigned as trans-4,cis-3,cis-3.
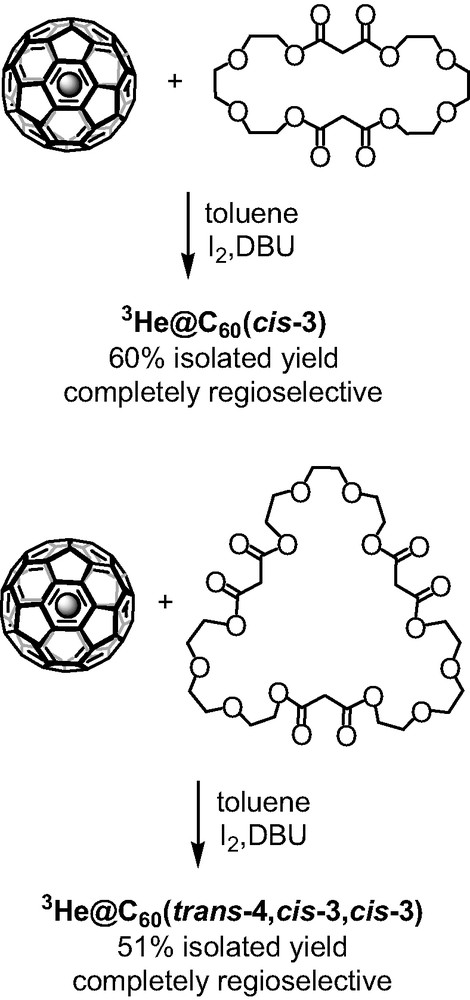
Functionalization of 3He@C60 with polyether malonates.
The successful synthesis of bis- and trisadducts of C60 utilizing the macrocyclic polymalonates prompted us to extend this study towards the preparation of enantiomerically pure polyadducts with an inherently chiral addition pattern. An addition pattern of a C60 adduct is inherently chiral when it belongs to one of the D3, C3, C2, or C1 point groups [3,8]. Fig. 3 shows the inherently chiral bisadducts with C2 symmetry and the inherently chiral trisadducts of C60 with C3 and D3 symmetry, as pairs of enantiomers. The chiroptical properties of fullerene derivatives with an inherently chiral addition pattern depend on the structure of the conjugated π system since, the whole chromophore itself is responsible for the chirality. Thus, such bulky building blocks represent attractive candidates for the construction of chiral macromolecular architectures such as dendrimers and spherical amphiphiles [9]. The utilization of opened-structure chiral bis-malonate tethers for the synthesis of enantiomerically pure bisadducts with an inherently chiral addition pattern was firstly reported by Nierengarten et al. [10]. In contrast, pure enantiomers of inherently chiral trisadducts of C60 have been synthesized by stepwise nucleophilic cyclopropanation of the [6,6] double bonds with diethyl bromomalonate and enantiomeric separation of the racemic mixture of the inherently chiral trisadducts by means of preparative chromatography (HPLC) on chiral stationary phases [11]. In addition, enantiomerically pure trisadducts of C60 were obtained by cyclopropanation of [6,6] bonds of the fullerene cage with C2-symmetrical bisoxazolines and subsequent HPLC chromatographic separation of the corresponding diastereomers on achiral stationary phases [12]. In these cases, only poor regio- and diastereoselectivities were observed, while tedious preparative HPLC chromatographic separations were required. To overcome these problems we addressed the concept of the cyclo-malonates targeting enantiomerically pure trisadducts of C60 and in a first step we performed the synthesis of an enantiomerically pure D3-symmetrical tris-malonate tether (Scheme 6). The macrocycle reacted cleanly with C60 in the presence of I2 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in toluene solvent, to afford three fullerene trisadducts (Scheme 6) [13]. The crude mixture was separated successfully by flash column chromatography on SiO2 followed by the spectroscopic characterization of the formed trisadducts. The minor adduct (1.4% isolated yield) was assigned as the trans-4,trans-4,trans-4 isomer which is inherently achiral. The two other trisadducts is a pair of diastereomers whose inherently chiral e,e,e addition pattern has an enantiomeric relationship. Their circular dichroism (CD) spectra showed almost perfect mirror-image behavior and pronounced Cotton effects. By direct comparison of the CD spectroscopic data, with those of enantiomerically pure [60]fullerene adducts with an e,e,e addition pattern, synthesized previously by non-tethered methods, the absolute configuration was unambiguously determined. Thus, the minor e,e,e trisadduct formed in 10.1% was assigned as the (+)-(R,R,R,R,R,R)-fC isomer, while the major (39.1%) as the (–)-(R,R,R,R,R,R)-fA. The formation of the e,e,e trisadducts is diastereoselective with a de value of 55% (HPLC) favoring the fA enantiomer.
Schematic representation of inherently chiral bis- (a) and trisadducts (b) of C60 as pairs of enantiomers. The bold red lines denote the [6,6] bonds carrying the addends and the blue motifs represent C2- or C3-symmetrical substructures with the C2 and C3 axes as symmetry elements.

Synthesis of the trans-4,trans-4,trans-4 and the enantiomerically pure (+)-(R,R,R,R,R,R)-fC-e,e,e and (–)-(R,R,R,R,R,R)-fA-e,e,e trisadducts.
The new approach applied for the tether-directed functionalization of C60 by the use of tethers containing the reactive malonate groups in a closed structure fulfilled our initial predictions and offers now a relatively simple way to access regioisomeric fullerene bis- and trisadducts with excellent selectivity. The regioselectivity can be controlled by tuning the length of the spacers connecting the malonate moieties, making this method highly flexible. The one step synthesis of the macrocyclic malonates via the condensation reaction of malonyl dichloride with an appropriate diol, combined with the high yield synthesis of fullerene multiadducts facilitates the wide applicability of the method. That was demonstrated by the synthesis of enantiomerically pure trisadducts of C60 having an inherently chiral addition pattern. Finally, the introduction of functional groups in the spacers will give the opportunity to synthesize novel fullerene architectures with promising properties as functional materials.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (Hi 468/14-1) for financial support.