

1 Introduction
Cycloaddition reactions are amongst the most important reactions for preparing functionalized fullerenes. Fullerenes are known to participate in a variety of cycloaddition reactions [1] including [1 + 2], [2 + 2], [3 + 2], [4 + 2], [6 + 2] and [8 + 2], but the [4 + 2] or Diels–Alder cycloaddition is by far the most studied and most useful fullerene cycloaddition reaction. A host of Diels–Alder dienes including traditional open chain 1,3-dienes, cyclic dienes, and heterodienes are known to react with [60]fullerene [2]. Like other Diels–Alder reactions, those involving [60]fullerene are believed to be concerted, stereospecific processes. They occur with complete regioselectivity such that the dienes add exclusively across 6,6 junctions on the [60]fullerene cage.
Due to its versatility, the Diels–Alder cycloaddition can be used to construct bis[60]fullerene and tris[60]fullerene adducts in which two or three fullerene moieties are held on a single molecule [3]. These exotic structures are beautiful synthetic targets but they also have interesting chemical and electronic properties. Thus, Belik et al. [4] used the two directional diene bis-o-quinodimethane to make a bis[60]fullerene adduct in which a flexible durene-type spacer separates the [60]fullerene moieties. The bis-o-quinodimethane diene was generated from two iodine induced 1,4-eliminations on 1,2,4,5-tetrakis(bromomethyl)benzene (Scheme 1).

Bis[60]fullerene adduct prepared by Belik et al. via reaction with bis-o-quinodimethane (see [4]).
Paquette and Graham [5] prepared novel bis[60]fullerene ‘ladder’ compounds in which the fullerene moieties are separated by repeating 1,4-cyclohexadienyl units. These molecules were prepared by reacting appropriate bifunctional dienes (Fig. 1) with C60 in boiling toluene (Scheme 2). A similar methodology was utilized to extend this chemistry in three directions. Paquette and Trego [6] used the bicyclic threefold diene hericene to generate a tris[60]fullerene–hericene adduct (Fig. 2) in 9% yield.
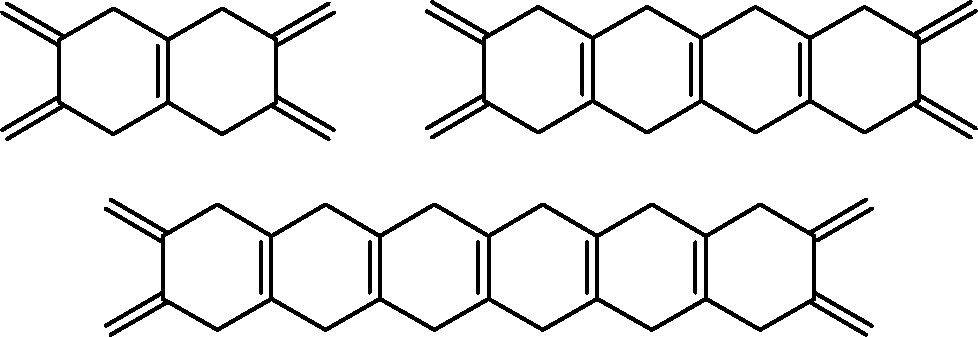
1,4-Cyclohexadienyl ladders: bifunctional dienes.
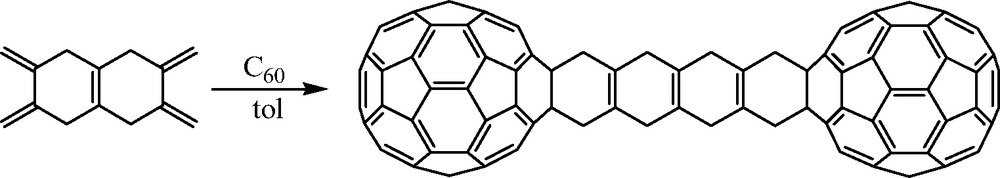
Bis[60]fullerene ‘ladder’ compound prepared by Paquette and Graham (see [5]).

Left: Hericene. Right: Hericene-tris[60]fullerene adduct prepared by Paquette and Trego (see [6]).
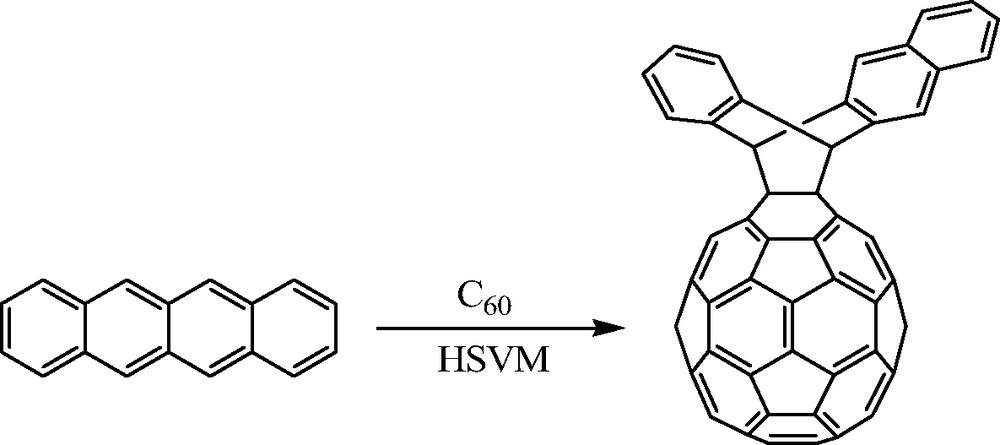
An interesting bis[60]fullerene adduct that places the two fullerene moieties in very close proximity to one another was reported by Murata et al. [7]. Using a solid-state high speed vibrational milling (HSVM) technique, the mechanochemical reaction between C60 and phthalazine was shown to produce an interesting bis[60]fullerene adduct in 14% yield (Scheme 3). The reaction proceeds through an initial [4 + 2] cycloaddition followed by loss of N2 in a retro-Diels–Alder reaction, followed by a second [4 + 2] cycloaddition of C60. Interestingly, this bis[60]fullerene adduct is not observed when the reaction is conducted in solution. Instead, an open cage fullerene hydrocarbon is formed.

Bis[60]fullerene adduct prepared by Murata et al. under HSVM conditions (see [7]).
We recognized the potential utility of using large acenes as platforms on which to cycloadd [60]fullerene and began pursuing the formation of bis[60]fullerene–acene and tris[60]fullerene–acene adducts in 1995. These studies have lead us to a greater understanding of the factors that influence Diels–Alder cycloadditions of [60]fullerene, especially inter- and intramolecular π–π stacking interactions between [60]fullerenes. In the process, we discovered several [60]fullerene Diels–Alder reactions that proceed with remarkable diastereoselectivity. The remainder of this review will focus on acenes and their Diels–Alder reactivity with [60]fullerene.
2 Acenes and Erich Clar
Acenes belong to the family of organic molecules known as polycyclic aromatic hydrocarbons (PAHs). Specifically, they are a subcategory of the PAH family in that they are alternant cata-annelated benzenoid ring systems [8]. In other words, acenes are a linear array of fused benzene rings, the smallest of which is the 2-ring system naphthalene. The name ‘acene’ was coined by the father of modern PAH chemistry, Clar [9]. The name is derived from one of the most common acenes, anthracene. Thus, the oft-used moniker ‘linear acene’ is redundant since the term ‘linear’ is already implied in Clar's definition.
Clar was born in Czech-Sudetenland and subsequently relocated to Glasgow in 1946. He was a Professor of Chemistry at Glasgow University from 1953 until 1972. Clar published over 170 research papers and monographs beginning in the late 1920s and continuing until the early 1980s. These contributions include the classic monograph Polycyclic Hydrocarbons which is still recognized today as an indispensable reference. Clar was posthumously awarded the first ‘Polycyclic Aromatic Hydrocarbon Research Award’ at the 11th International Symposium on Polynuclear Aromatic Hydrocarbons, September 24, 1987. He died just a few days before news of the award reached his home in 1987. Without question, Clar's contributions toward acene syntheses paved the way for today's numerous applications. Indeed, acenes possess a set of physical and chemical properties that make them interesting components of oriented thin films [10], organic light emitting diodes (LEDs) [11], organic field-effect transistors (FETs) [12], liquid crystals with enhanced electron transport [13], and single crystal semi-conductors [14]. They also exhibit interesting chemistries including their propensity to cycloadd [60]fullerene in syn-diastereoselective fashion (vide infra). The known acenes are shown in Fig. 3 with all but octacene having been prepared by Clar. While larger acenes are easily envisioned, these compounds have never been successfully prepared and characterized due mostly to their photo-instability, high reactivity, and poor solubility.

Structures and names of the known acenes.
3 [60]Fullerene–acene chemistry
The first [60]fullerene–acene reaction studied was that between [60]fullerene and anthracene. Schlueter et al. [15] reported that upon boiling a toluene solution of anthracene and [60]fullerene for 3 days, the corresponding [60]fullerene–anthracene monoadduct is formed and isolated in 13% yield. The [60]fullerene–anthracene monoadduct is not a thermally stable molecule, a characteristic highlighted by Tsuda et al. [16] who observed a temperature dependence on the yield of monoadduct formed. Thus, for reactions run in toluene solution at 50, 80 and 100 °C, [60]fullerene–anthracene monoadduct yields of 24%, 17% and 8.5%, respectively, were observed. The reaction is reversible (Scheme 4) with reactants favored at higher temperatures. In practice, one observes that isolated [60]fullerene–anthracene monoadduct begins to thermally degrade at approximately 60 °C.

Reversible Diels–Alder reaction between C60 and anthracene (see [16–18]).
Komatsu and co-workers studied the reaction between anthracene and [60]fullerene under HSVM conditions producing the [60]fullerene–anthracene monoadduct in 55% yield. When tetracene and [60]fullerene are reacted under HSVM conditions, the corresponding [60]fullerene–tetracene monoadduct (Scheme 5) is produced in 61% yield [17].
Mechanochemical synthesis of a [60]fullerene–tetracene monoadduct by Komatsu and co-workers (see [18]).
Sarova and Berberan-Santos [18] studied the kinetics of [60]fullerene cycloaddition across anthracene and tetracene, as well as the kinetics of the corresponding retro-Diels–Alder reactions. Tetracene cycloadds faster than anthracene and the corresponding [60]fullerene-tetracene monoadduct is slower to undergo a retro-Diels–Alder reaction. Larger acenes are long known to react faster than smaller acenes in Diels–Alder reactions [19]. Likewise, pentacene reacts noticeably faster with [60]fullerene than does tetracene and the corresponding [60]fullerene–pentacene monoadduct (vide infra) is less inclined to decompose via a retro-Diels–Alder reaction.
Herranz and Echegoyen [20] studied the thermal and electrochemical stability of the Diels–Alder adducts prepared upon reacting [60]fullerene with 9-anthracenylmethyl ethyl malonate and di-9-anthracenylmethyl malonate (Scheme 6). While both adducts showed signs of thermal degradation at temperatures as low as 50 °C, the adduct of 9-anthracenylmethyl ethyl malonate was shown to be stable to electroreductive (controlled potential electrolysis) conditions. Base promoted Bingel reactions involving the adduct of 9-anthracenylmethyl ethyl malonate lead to multiple products including both intramolecular cyclopropanation product (Scheme 6) and some dimeric species (i.e. products of intermolecular reactions).

Diels–Alder adducts prepared by Herranz and Echegoyen upon reacting [60]fullerene with 9-anthracenylmethyl ethyl malonate (top) and di-9-anthracenylmethyl malonate (bottom). See [20].
While a lack of thermal stability in [60]fullerene-anthracene adducts can be problematic in some applications, it can be advantageous in others. Wang suggested the use of silica or resin-supported 9-substituted anthracenes for the separation of fullerenes from complex mixtures. The concept has yet to be demonstrated, but Wang did study the retro-Diels–Alder rates for three [60]fullerene-anthracene monoadducts using 9-substituted anthracenes as diene [21]. Not surprisingly, these adducts decompose at relatively low temperatures (i.e. < 60 °C). A suitably bulky 9-substituted or 9,10-disubstituted anthracene may undergo facile retro-Diels–Alder reactions at or near room temperature, enabling a resolution of fullerenes on modified silica or resin.
Indeed, Hirsch and co-workers successfully exploited the thermal reversibility of the Diels–Alder reaction between [60]fullerene and 9,10-dimethylanthracene (DMA) for synthetic gain. Noting regioselective formation of equatorial (e) bisadducts and e,e-trisadducts in Bingel addition chemistries (i.e. base promoted cyclopropanations via bromomalonates), Hirsch demonstrated that adding an excess of DMA to [60]fullerene creates an equilibrium mixture containing e-C60(DMA)2 and e,e-C60(DMA)3 templates that direct subsequent cycloadditions to the remaining equatorial sites [22]. The method allows the synthesis of a number of Bingel-type Th symmetric hexakisadducts, C66(COOR)12 (Scheme 7), in yields up to 50%. Thus, the reversible addition of DMA promotes regioselective (irreversible) cyclopropanation in the presence of bromomalonate and DBU leading ultimately to Th symmetric hexakisadducts in which all equatorial sites are cyclopropanated.
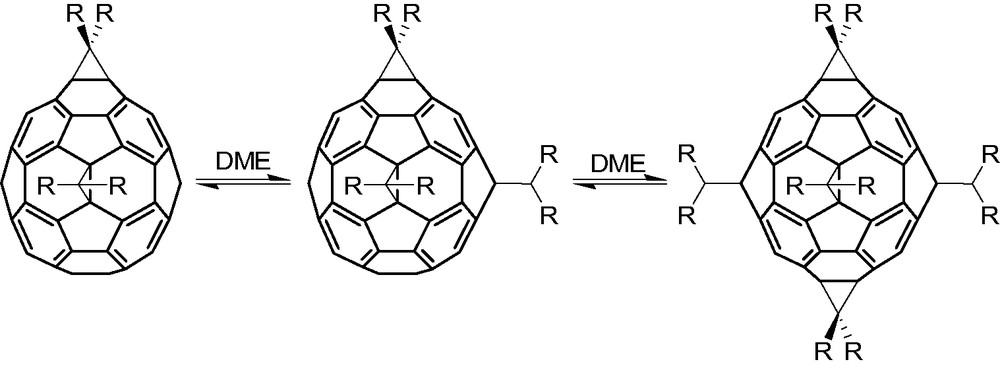
Formation of a Bingel-type Th symmetric hexakisadduct (right, R = CO2R′) by Lamparth et al. using excess DMA as reversible templating agents. See [22]. The sixth cyclopropanation is not seen in this view of the hexakisadduct.
Kräutler et al. [23] discovered that heating an X-ray quality crystal of the [60]fullerene-anthracene monoadduct to 180 °C for 10 min produced a 1:1 mixture of the antipodal (trans-1) [60]fullerene–bisanthracene adduct (Fig. 4) and C60 in 48% yield. They attribute this unique solid-state reaction to crystal stacking of the [60]fullerene–anthracene monoadduct molecules which places the anthracene moiety of one molecule in close proximity to the antipodal (trans-1) position of an adjacent [60]fullerene–anthracene monoadduct. Heating the crystal induces retro-Diels–Alder reactions producing ‘free’ anthracene molecules in the crystal lattice. The freed anthracene molecules can cycloadd to the same [60]fullerene from which they came or alternatively to an adjacent [60]fullerene-anthracene monoadduct. In this latter instance, a trans-1 [60]fullerene-bisanthracene adduct molecule is produced. Although the reaction is limited in scope, Kräutler's method represents one of the very few options available to fullerene chemists to prepare a trans-1 bisadduct in reasonable yield. Kräutler et al. [24] was successful in extending this methodology to a number of different substituted anthracenes.
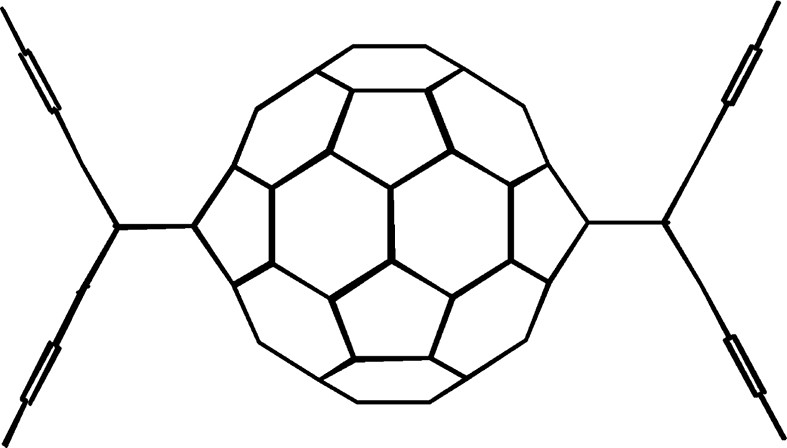
Kräutler’s antipodal (trans-1) [60]fullerene-bisanthracene adduct (see [23]).
Interested in the possibility that two [60]fullerenes could cycloadd across a single pentacene backbone, Mack and Miller [25] studied the reaction between pentacene and [60]fullerene in boiling toluene. Under these conditions, a C2v symmetric [60]fullerene–pentacene monoadduct, 1, forms in 54% yield (Scheme 8), a result of [60]fullerene cycloaddition across the central 6,13 carbons of the pentacene backbone. Using a fivefold excess of [60]fullerene, the yield of C2v 1 can be increased to 90%. The reaction is highly regioselective and shows no evidence for formation of either a Cs symmetric monoadduct (i.e. resulting from addition across the 5,14 carbons of the pentacene backbone), or the cis or trans-bis[60]fullerene–pentacene adducts, 2 and 3, respectively (Fig. 5), as sought by Kräutler et al. [24]. An X-ray crystal structure (Fig. 6) of 1 reveals [26] a similar nested crystal packing as reported by Kräutler and co-workers for the [60]fullerene-anthracene monoadduct crystal [23]. Thus, each [60]fullerene moiety nests inside the pentacene arms of an adjacent [60]fullerene-pentacene monoadduct molecule.
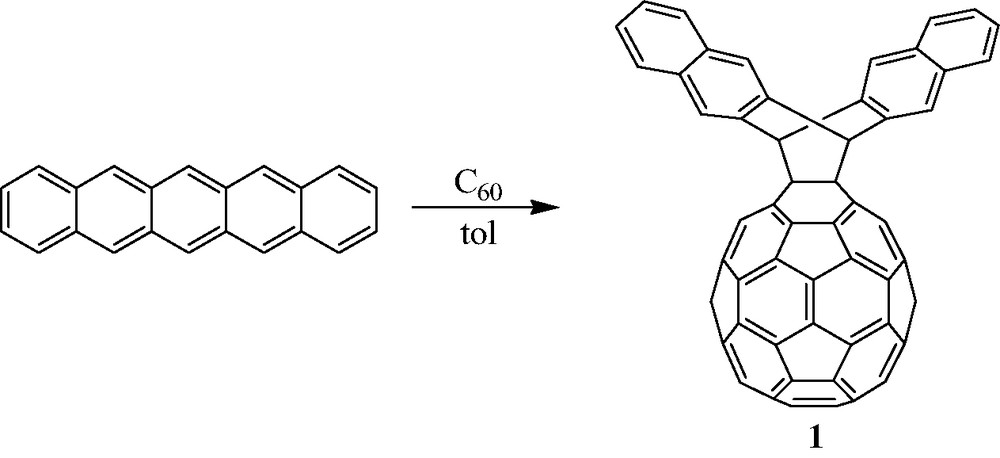
Solution phase Diels–Alder cycloaddition between [60]fullerene and pentacene proceeds regioselectively across the central ring of pentacene (see [25]).

Cis- and trans-bis[60]fullerene-pentacene adducts, 2 and 3, respectively, sought by Mack and Miller (see [25]).
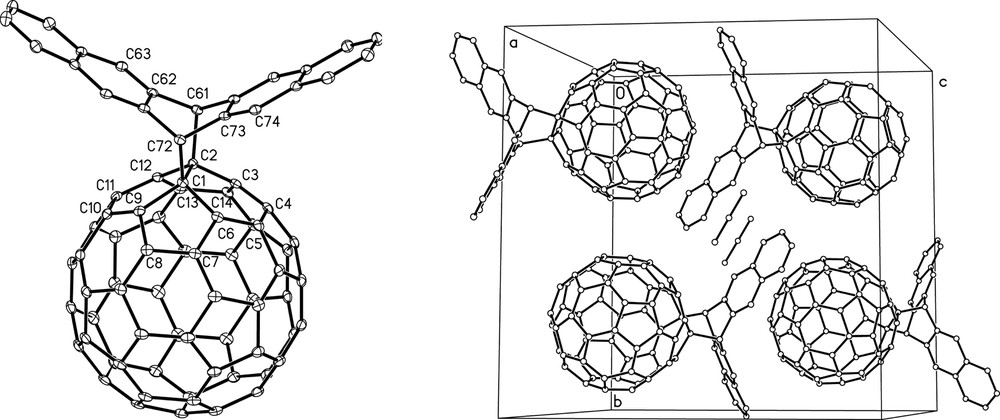
X-ray crystal structure of C2v [60]fullerene-pentacene monoadduct 1 (reprinted with permission from [26]).
Murata et al. [17] subsequently demonstrated that a bis[60]fullerene adduct of pentacene could be prepared in low yield using the HSVM technique. Under mechanochemical conditions, C2v 1 was isolated in 19% yield along with a bis[60]fullerene-pentacene adduct in 11% yield. Murata et al. assigned an anti-stereochemistry (i.e. 3) to the bis[60]fullerene-pentacene adduct prepared under HSVM conditions, but this assignment is likely in error (vide infra).
4 Diastereoselective [60]fullerene cycloadditions across 6,13-disubstituted pentacenes
Formation of either 2 or 3 requires initial cycloaddition of [60]fullerene across the 5,14 carbons of pentacene to yield a Cs symmetric monoadduct. However, semi-empirical calculations reveal that 6,13 cycloaddition across pentacene is both kinetically and thermodynamically preferred to 5,14 cycloaddition [27]. Likewise, the probability of forming 2 and 3 in good to excellent yield from [60]fullerene and pentacene is remote. Consequently, we sought new reactions where the preference for 6,13 cycloaddition across a pentacene backbone would be reduced. PM3 calculations indicate that phenyl substituents at the 6,13 positions of pentacene are sufficiently large as to bias the energetics such that 5,14 cycloaddition is both kinetically and thermodynamically preferred (Fig. 7). Surprisingly, a relatively large kinetic bias already exists for the cycloaddition of the small dienophile ethylene. With a larger dienophile like bicyclopentylidene, the kinetic and thermodynamic bias towards 5,14 cycloaddition is truly impressive. Guided by these calculations, we prepared 6,13-diphenylpentacene and reacted it with excess [60]fullerene (Scheme 9). Remarkably, the reaction proceeds with complete syn diastereoselectivity leading to the C2v symmetric cis-bis[60]fullerene-diphenylpentacene adduct, 4, in 85% isolated yield [27]. The syn diastereoselectivity was originally deduced by a careful examination of 1H and 13C NMR spectra. For example, the 1H NMR spectrum for C2v 4 reveals a set of 5 phenyl 1H signals (two quasi-doublets and three quasi-triplets) for the phenyl substituents that rotate slowly on the NMR timescale (Fig. 8). These signals cluster around the expected AA′MM′ pattern for the remaining aromatic protons on the pentacene backbone. Had the C2h trans-bis[60]fullerene-diphenylpentacene adduct formed, only three 1H NMR signals (one quasi-doublet and two quasi-triplets) would have been observed for the phenyl substituents, regardless of their rate of rotation.

PM3 semi-empirical calculations reveal a kinetic and thermodynamic bias for 5,14 cycloaddition across 6,13-diphenylpentacene (see [27]).

Diastereoselective synthesis of cis-bis[60]fullerene-diphenylpentacene adduct, 4, by Miller and Mack (see [27]).

1H NMR spectrum of cis-bis[60]fullerene-diphenylpentacene adduct, 4, prepared by Miller and Mack (see [27]).
An X-ray crystal structure (Fig. 9) was subsequently obtained [26] for 4 confirming the NMR structural characterization. The crystal structure reveals that two 5-membered rings on adjacent fullerene moieties directly face each other but are not quite parallel to one another. Due to the constraints provided by the pentacene backbone, these abutting rings have an eclipsed orientation. The carbon atoms that make the closest contact are 3.065(8) Å apart and lie at the top of the nearest neighbor 5-membered rings, furthest removed from the pentacene backbone. The distance between the centers of the abutting pentagons is 3.284 Å. The centroid-to-centroid distance between adjacent fullerenes is 9.805 Å. For comparison, the analogous centroid-to-centroid distance in pristine [60]fullerene, which is orientationally disordered, is 9.94 Å at 110 K [28] while the corresponding distances in C60·4C6H6 [29] and in 2C60·3CS2 [30], in their low temperature ordered phases are 9.8722 Å (90 K) and 9.9217 Å (93 K), respectively. In none of these structures do pentagons directly face each other in an eclipsed fashion. The cis-bis[60]fullerene adduct 4 shows excellent solubility in common organic solvents including chloroform. This solubility must be due in large part to the phenyl substituents on the pentacene backbone as the bis[60]fullerene-pentacene adduct prepared under HSVM conditions exhibits poor solubility [17].
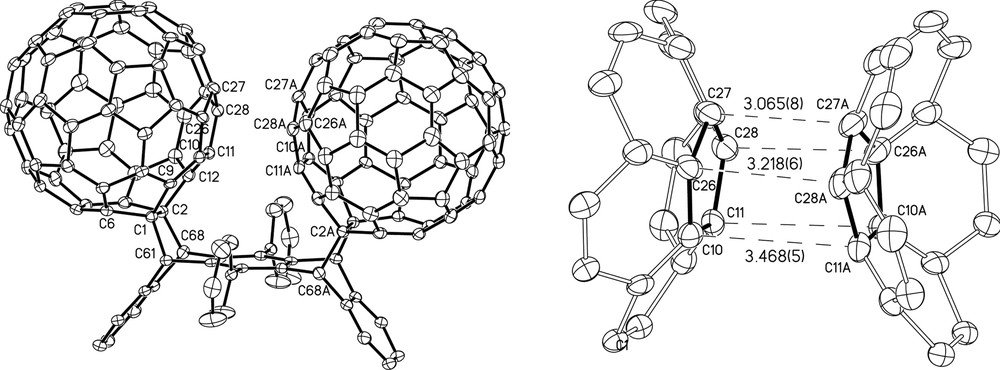
X-ray crystal structure for cis-bis[60]fullerene-diphenylpentacene adduct, 4 (reprinted with permission from [26]).
Although readily synthesized and isolated, 6,13-diphenylpentacene is photolytically unstable. It readily adds oxygen in solution in the presence of ambient light. For long-term storage, it is best to store 6,13-diphenylpentacene dry and in the dark. When stored in this manner, it is stable for months without significant decomposition. In addition to 6,13-diphenylpentacene, a variety of other 6,13-disubstituted pentacenes were synthesized [31] as illustrated in Fig. 10.

Several 6,13-disubstituted pentacenes synthesized and reacted with [60]fullerene by Miller, Mack and Briggs (see [31]).
Thus, a fivefold excess of [60]fullerene was reacted with 6,13-di(4′-hydroxymethylphenyl)pentacene, 5, in CS2 to produce the corresponding cis-bis[60]fullerene-di(hydroxymethylphenyl)pentacene adduct, 8, in 75% isolated yield [27]. Hindered rotation of the hydroxymethylphenyl substituents once again allowed for complete stereochemical characterization, the product 8 forming in a highly syn-diastereoselective fashion (Fig. 11).
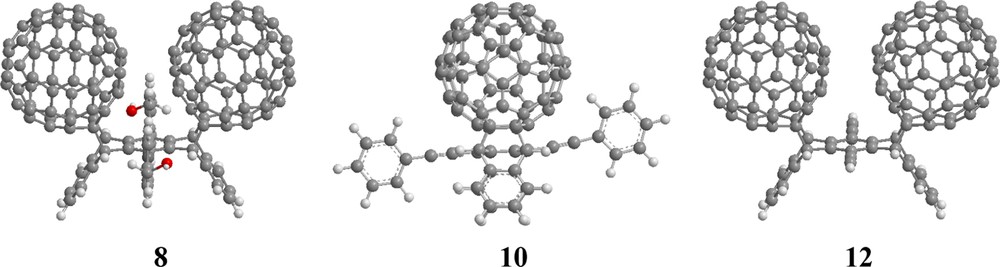
Structures of bis[60]fullerene-acene adducts formed upon reacting [60]fullerene with 6,13-disubstituted pentacenes 5–7. Compound 10 is viewed along the long-axis of the pentacene backbone (see [27,31]).
Similar conditions were utilized for the reaction between [60]fullerene and 6,13-diethynylphenylpentacene, 6, to produce the corresponding Cs symmetric [60]fullerene-diethynylphenylpentacene monoadduct, 9, in 83% isolated yield. In this reaction, the corresponding bis[60]fullerene–diethynylphenylpentacene adduct, 10, is formed in only 14% isolated yield [32]. The sluggishness of the reaction with 6 is attributed to the alkynyl substituents. The electronegative sp hybridized carbons cause the pentacene backbone to be less electron rich and therefore less reactive in the Diels–Alder reaction. Due to the acetylene spacer, hindered rotation of the phenyl groups is not observed in this system. Consequently, the 1H and 13C NMR spectra do not distinguish between cis- and trans-bis[60]fullerene adducts. It is nonetheless certain that only one diastereomer forms in a highly selective reaction. Given the results discussed above, the product is likely to possess a cis-stereochemistry (Fig. 11).
Diethynylpentacene, 7, was also reacted with [60]fullerene. Since 7 is very unstable, it was generated and used as needed from 6,13-di(trimethylsilylethynyl)pentacene. Reaction of 6,13-di(trimethylsilylethynyl)pentacene with TBAF in chloroform followed by immediate purification on a short silica column affords a blue solution of 7 which was added directly to a solution of [60]fullerene in CS2. In this manner, the Cs symmetric [60]fullerene–6,13-diethynylpentacene monoadduct 11 was isolated in 2.2% yield while the bis[60]fullerene-6,13-diethynylpentacene adduct 12 was isolated in 8% yield [32]. These low isolated yields are attributed to both the reduced reactivity of 7 as well as difficulties in purifying sparingly soluble 11 and 12.
5 A tris[60]fullerene adduct of 6,8,15,17-tetraphenylheptacene
We endeavored to extend our [60]fullerene-acene methodology to larger acene systems such as 6,8,15,17-tetraphenylheptacene, 13. Acene 13 is predicted to be highly reactive and unstable. It was prepared and used in situ upon reacting the stable, H-protected 5,7,9,14,16,18-hexahydro-6,8,15,17-tetraphenylheptacene, 14 (Scheme 10), with DDQ in the presence of excess [60]fullerene to afford the cis,cis-tris[60]fullerene-tetraphenylheptacene adduct 15 (Scheme 11) in 74% crude and 20% isolated yield [33]. The cis,cis-tris[60]fullerene adduct 15 is the only product isolated in the reaction. The positive assignment of a cis,cis-stereochemistry for 15 was enabled by MM2 calculations and a careful analysis of NMR data. Similar to the cases with cis-bis[60]fullerene adducts 4 and 8, the stereochemical assignment for 15 was assisted by the slow rotation of phenyl substituents on 15.

Synthesis of 5,7,9,14,16,18-hexahydro-6,8,15,17-tetraphenylheptacene, 14, by Miller and Briggs (see [33]).
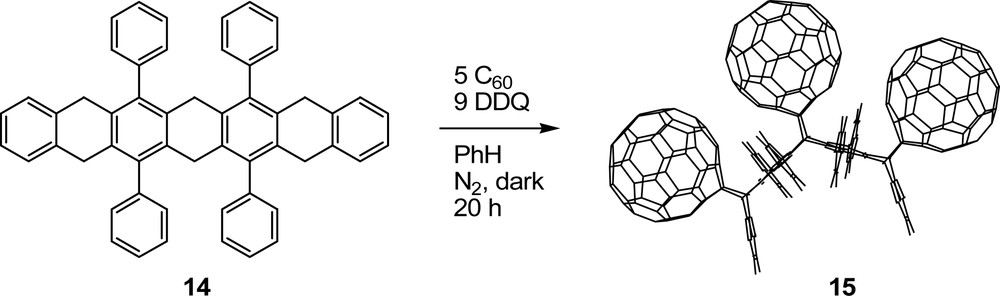
Synthesis of cis,cis-tris[60]fullerene-tetraphenylheptacene adduct, 15, by Miller and Briggs (see [33]).
Given all of the data collected for reactions between [60]fullerene and 6,13-disubstituted pentacenes and 6,8,15,17-tetraphenylheptacene, there can be no doubt that [60]fullerenes prefer to cycloadd across large acenes in a syn-diastereoselective fashion. But what is the origin of this diastereoselectivity?
6 Why do [60]fullerenes add across large acenes in a syn diastereoselective fashion?
The origin of the syn-diastereoselective addition of [60]fullerenes across large acenes is favorable van der Waals or π–π stacking interactions between adjacent [60]fullerene moieties. There is ample experimental and computational evidence to support this conclusion.
The X-ray crystal structure of cis-bis[60]fullerene-diphenylpentacene adduct, 4, illustrates that the distance between the centers of the abutting pentagons on adjacent [60]fullerene moieties is 3.284 Å (Fig. 9). This distance is only slightly shorter than the 3.35 Å van der Waals spacing between adjacent sheets of planar graphite. Moreover, this is the approximate distance between the carbon layers of multi-walled carbon nanotubes (MWNTs), and between individual single-walled carbon nanotubes (SWNTs) that have bundled into rope-like structures. Could these similar clustering effects involving graphite, [60]fullerene, MWNTs and SWNTs – all different allotropes of sp2 hybridized carbon – be a matter of coincidence? No. In all cases, the ‘clusters’ are stabilized by favorable π–π stacking interactions between the nanostructured carbon forms.
A molecular mechanics (MM2) calculated ground state [34] for 2 is in excellent agreement with the X-ray crystal structure [26] for 4. In the MM2 ground state structure, the distance between the centers of the abutting pentagons on adjacent [60]fullerene moieties is 3.3 Å. Nearest neighbor pentagons should be slightly closer in the syn transition states preceding 2 and 4, and indeed a PM3–MM2 hybrid transition state structure reveals a distance of 3.2 Å. It is interesting and informative to note that the semi-empirical methods (e.g. AM1, PM3, MNDO, etc.) fare poorly in predicting the structures of cis-bis and cis,cis-tris[60]fullerene–acene adducts because, unlike MM2, they are not parameterized to account for van der Waals interactions. That is, the MM2 calculated ground state for 4 matches the crystal structure so well because the MM2 force field specifically accounts for van der Waals interactions between adjacent [60]fullerene moieties.
When [60]fullerenes add across large acenes, the reactions must proceed through initial formation of a [60]fullerene-acene monoadduct. Once formed, the first [60]fullerene addend effectively directs subsequent [60]fullerene additions across the same (syn) face of the acene via [60]fullerene–[60]fullerene π–π stacking interactions. The unique directing effect of [60]fullerene addends is exemplified by a study [34] of the reaction between 6,13-diphenylpentacene and the small dienophile dimethyl acetylenedicarboxylate (DMAD). Using a 20-fold excess of DMAD in boiling toluene, a 1:1.3 mixture of cis and trans-bisDMAD–diphenylpentacene adducts form in quantitative yield (Scheme 12). The slight preference for the trans-bisDMAD–diphenylpentacene adduct is likely due to a modest steric effect. Most importantly, the absence of syn diastereoselectivity in the DMAD reaction suggests that a special interaction must exist between adjacent [60]fullerene moieties in the [60]fullerene reaction.

Reaction between 6,13-diphenylpentacene and DMAD produces a 1:1.3 mixture of cis and trans-bisDMAD-diphenylpentacene adducts (see [34]).
Because even slight deformations from ideal π–π stacking geometries can impact binding energies [35] (stabilities), we reasoned that sterically demanding 6,13-substituents on pentacene should diminish the syn diastereoselectivity of [60]fullerene addition. We successfully achieved steric disruption of [60]fullerene–[60]fullerene π–π stacking interactions in a syn transition state leading to a cis-bis[60]fullerene–acene adduct by utilizing the bulky acene 6,13-di(trimethylsilylethynyl)pentacene [34]. Computationally, 6,13-di(trimethylsilylethynyl)pentacene, 16, represents a reasonable compromise between 6,13-diphenylpentacene, which exhibits excellent reactivity, and a highly hindered acene like bis(trimethylsilyl)pentacene that would seemingly retard Diels–Alder reactivity. A PM3-MM2 hybrid syn transition state structure for the cis-bis[60]fullerene-di(trimethylsilylethynyl)pentacene adduct suggested mild steric disruption without a complete loss in reactivity. Likewise, 16 (Fig. 12) was prepared and reacted with a fivefold excess of [60]fullerene to produce a 2.5:1 ratio of bis[60]fullerene-di(trimethylsilylethynyl)pentacene adducts in an overall sluggish reaction [34]. The starting acene lacks suitable stereochemical handles such that we cannot distinguish cis and trans product isomers, but the formation of a mixture of diastereomers rather than a single product suggests that the trimethylsilylethynyl substituents do indeed disrupt [60]fullerene–[60]fullerene π–π stacking interactions.
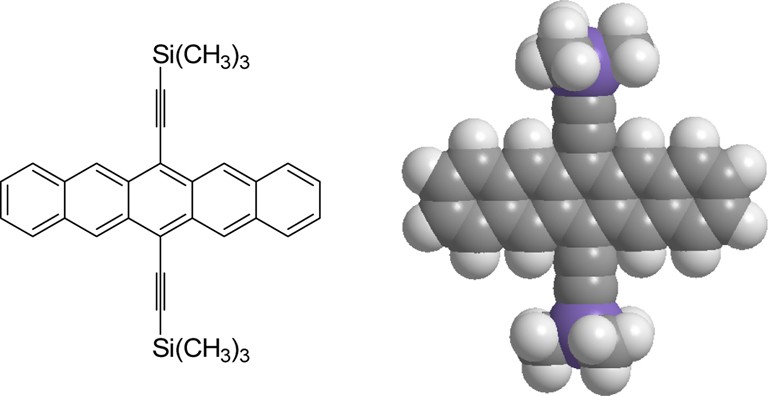
The sterically hindered di(trimethylsilylethynyl)pentacene, 16 (see [34]).
We further probed the spatial requirements for [60]fullerene–[60]fullerene π–π stacking interactions by studying the reaction between [60]fullerene and 6,8,15,17-tetraphenylheptacene–7,16-quinone, 17. Quinone 17 (the penultimate structure of Scheme 10) is unique in that it possesses two reactive anthracene moieties separated by a quinone ring. It is the central ring of each anthracene moiety that is reactive with [60]fullerene. These central rings are sufficiently far removed from one another that [60]fullerene-[60]fullerene π–π stacking interactions are negligible in the syn-transition state. Calculations reveal the spatial separation of nearest neighbor carbon atoms in the syn-transition state to be nearly 7 Å, well beyond the ideal 3.2–3.4 Å value for [60]fullerene–[60]fullerene π–π stacking. Upon reacting 17 with excess [60]fullerene, a 1:1.1 mixture of bis[60]fullerene adducts 18 and 19 form (Fig. 13) with the trans structure slightly preferred [36]. The lack of syn-diastereoselectivity in this reaction contrasts sharply with the highly diastereoselective syn-addition of [60]fullerenes across 6,13-diphenylpentacene and 6,8,15,17-tetraphenylheptacene. Without question, the presence or absence of syn-diastereoselectivity in these reactions is closely linked to spatially dependent [60]fullerene–[60]fullerene π–π stacking interactions. While the Diels–Alder reactive sites on 6,13-diphenylpentacene and 6,8,15,17-tetraphenylheptacene are ideally spaced to accommodate [60]fullerene–[60]fullerene π–π stacking, those on quinone 17 are not.
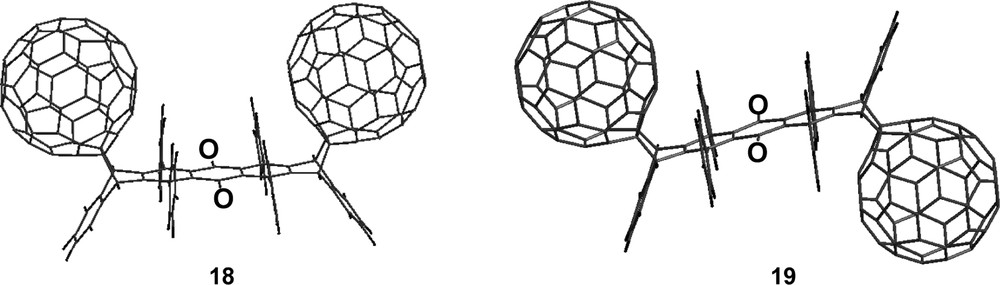
Reaction between 6,8,15,17-tetraphenylheptacene-7,16-quinone, 17, and [60]fullerene produces a 1:1.1 mixture of cis and trans-bis[60]fullerenetetraphenylheptacenequinone adducts, 18 and 19, respectively (see [36]).
Finally, it is interesting to note that bis[60]fullerene adducts 18 and 19 undergo facile retro-Diels–Alder reactions at temperatures marginally above 25 °C [36]. These adducts are even less stable than [60]fullerene–anthracene monoadducts. Compounds 4 and 15 on the other hand are thermally stable to temperatures in excess of 100 °C. The increased thermal stability of 4 and 15 can be largely attributed to the stabilizing nature of [60]fullerene–[60]fullerene π–π stacking interactions.
7 Conclusions and future direction
Cycloaddition reactions between acenes and [60]fullerene produce beautiful molecules with highly interesting structures. Because large acenes can accommodate more than one [60]fullerene cycloaddition, bis and tris[60]fullerene adducts can be prepared. These reactions become facile once suitable directing substituents (e.g. phenyl groups) are added to the acene. In all cases, [60]fullerenes add in a syn-diastereoselective fashion due to favorable π–π stacking interactions between adjacent [60]fullerene moieties. In the future, the chemical, electrochemical and physical properties of [60]fullerene-acene adducts already prepared will be investigated. Moreover, the methodology will be extended to larger acenes such that tetrakis, pentakis, and even hexakis[60]fullerene-acene adducts can be prepared.
Acknowledgements
The authors thank the National Science Foundation (NER 0403954 and NSEC 0425826) for partial support of the [60]fullerene-acene research performed in our laboratories.