

1 Introduction
The regio- and, if applicable, the stereoselective preparation of multiadducts of fullerenes is of prime importance for the preparation of functional fullerene derivatives and fullerene-based advanced materials [1]. This is related to the fact that the properties of the residual fullerene chromophore of derivatives critically depend on the functionalization pattern and that their geometry plays a crucial role in the relative arrangement of components, thereby determining the structure of such materials. Successive addition of independent reagent molecules to fullerenes generally leads to a mixture of regio- and/or stereoisomers, which are difficult to separate, and the desired compound may not even be formed at all [2,3]. In order to gain regio- and stereocontrol over bis- and multiadditions to fullerenes, Diederich and co-workers first applied the tether-directed remote functionalization [4] to the carbon spheroids in 1994 [5]. It consists in the attachment to the cage molecules of an anchor carrying a reactive group at the end of a tether (Fig. 1). Conformational preferences of the spacer and steric constraints ideally allow the reactive group to attack only a single, specific position within the molecular conjugate. In some cases, it has been possible to reduce the role of the tether to that of an auxiliary, which is removed after achievement of a selective bis- or multiaddition and is, therefore, termed a template [6]. The tether-directed remote functionalization has become the most generally applicable method for selective multiadditions to fullerenes [7,8]. The present account gives an overview of its development in the Diederich group over the past 11 years.
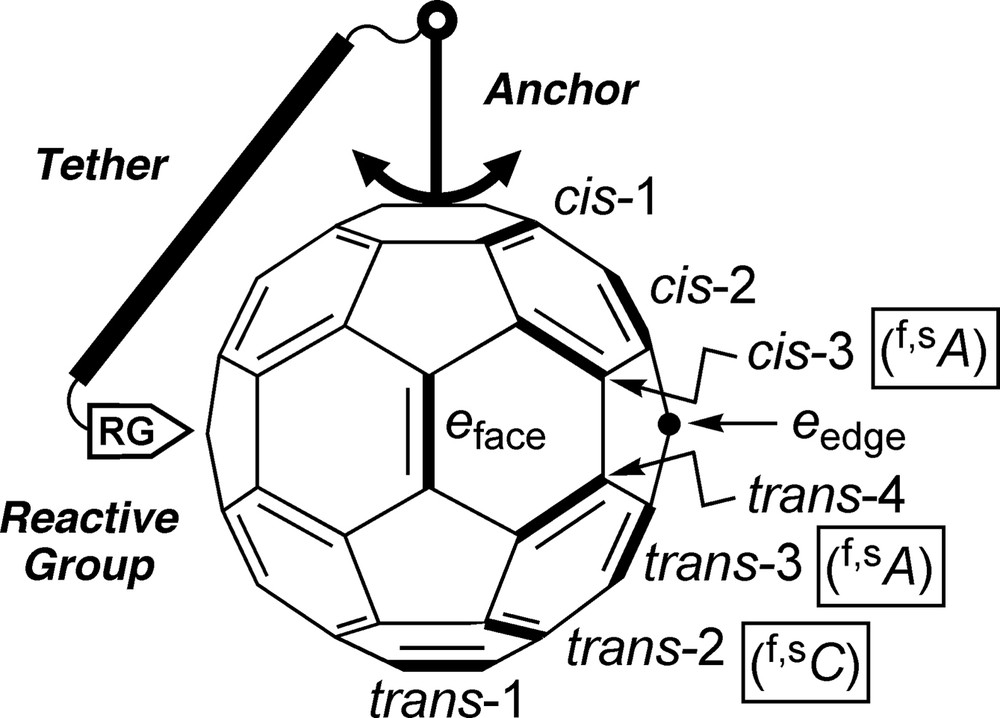
Illustration of the concept of tether-directed remote functionalization of C60, trivial nomenclature of possible bisadduct isomers [10], and stereodescriptors for the highlighted inherently chiral addition patterns [9,12].
2 Isomerism of [60]fullerene bisadducts
Functionalization of one of the 30 identical 6–6 bonds (common edges of two six-member rings) of Ih-C60 [2,3] by addition of a C2v-symmetric addend (e.g. anchor in Fig. 1) lowers the symmetry of the derivatized carbon cage to C2v, thereby eliminating the equivalence of the remaining 6–6 bonds. As a result, addition of a second, identical residue can, in principle, lead to eight different bisadduct regioisomers. They can be unambiguously named according to IUPAC (International Union of Pure and Applied Chemistry) nomenclature [9] or, in a more intuitive way, by indication of the relative arrangement of the addends as proposed by Hirsch et al. [10] (Fig. 1): the two addends can be located in the same hemisphere (cis) or in opposite hemispheres (trans) of the carbon cage and, in addition, there is a borderline case which is termed equatorial or e. There are three possible arrangements for two addends within a hemisphere (cis-1, cis-2, and cis-3) and four of them involving both hemispheres (trans-1, trans-2, trans-3, and trans-4). If two different, C2v-symmetrical addends are considered, the structural degeneracy of e arrangements vanishes and the second addend can occupy two different e sites (eface and eedge, Fig. 1) in relation to the first. This leads to a total of nine possible constitutional bisadduct isomers.
The number of bisadducts further increases if stereoisomers are included. In fact, the cis-3, trans-3, and trans-2 addition patterns are inherently chiral, that is, the corresponding functionalization with any two addends (identical or different, achiral or chiral) provides a chiral molecule [11–13]. Other addition patterns (cis-1, cis-2, and trans-4) become chiral only with two different addends: they are termed noninherently chiral. As to the remaining functionalization patterns, trans-1 bisadducts are always achiral unless the addends include appropriate stereogenic units. In the case of e bisadducts with two identical, Cs-symmetrical methano-type addends, i.e., carrying two different substituents at the bridging C-atoms – the methano C-atom in eface position is a stereogenic center but the addition pattern remains achiral [13]. In other cases of e bisadducts with identical, Cs-symmetrical addends, the addition pattern becomes noninherently chiral without new stereogenic centers appearing in the addends [13]. The situation in e bisadducts depends critically on structural details of the addends and shall not be discussed here exhaustively.
As recommended by the IUPAC, the configuration of a fullerene derivative with a chiral functionalization pattern is specified by the stereodescriptors (f,sC) and (f,sA) (f = fullerene, s = systematic [numbering], C = clockwise, A = anticlockwise) [9,12]. C and A correspond to the direction of an arc traced from C(1) via C(2) to C(3) in a fullerene derivative numbered according to IUPAC [9,14]. Observation of the rule of the lowest set of locants for the addends is of particular importance in the numbering procedure. Fig. 1 displays the stereodescriptors for the highlighted examples of inherently chiral cis-3, trans-3, and trans-2 addition patterns.
3 The tether-directed remote functionalization of C60: early developments
Although the various formal double bonds of a [60]fullerene monoadduct show somewhat different reactivities [3,15], selectivity in bisadduct formation is generally limited and regio- or stereoisomeric product mixtures result. These are often difficult to purify and, furthermore, the desired isomer may only be formed in minor amounts, if at all. Trying to remedy this unsatisfactory situation, the Diederich group tested the tether-directed remote functionalization [4] as a rational approach towards selectively multifunctionalized fullerenes that are difficult to obtain in thermodynamically or kinetically controlled successive additions of unconnected reagents [5,16]. The first step of this method consists in the attachment to the fullerene of an anchor carrying a reactive group at the end of a tether (Fig. 1). As a result of steric and conformational constraints imposed by anchor and tether, the reactive group should ideally add to a single, precise position of the carbon spheroid.
Anchor–tether–reactive group conjugates such as 1 (Scheme 1) were designed by PM3 semiempirical calculations to afford the desired multiadduct with the lowest relative heat of formation [5,16]. Indeed, fixation of the anchor (malonate group of 1) to the fullerene core by nucleophilic cyclopropanation in the presence of a non-nucleophilic base (Bingel reaction [17]) and subsequent addition of the reactive buta-1,3-diene groups by intramolecular Diels–Alder reactions under high dilution conditions afforded the predicted trisadduct 2 with complete regioselectivity [5,16]. Its e,e,trans-1 addition pattern is not attainable through successive additions of untethered reagents.
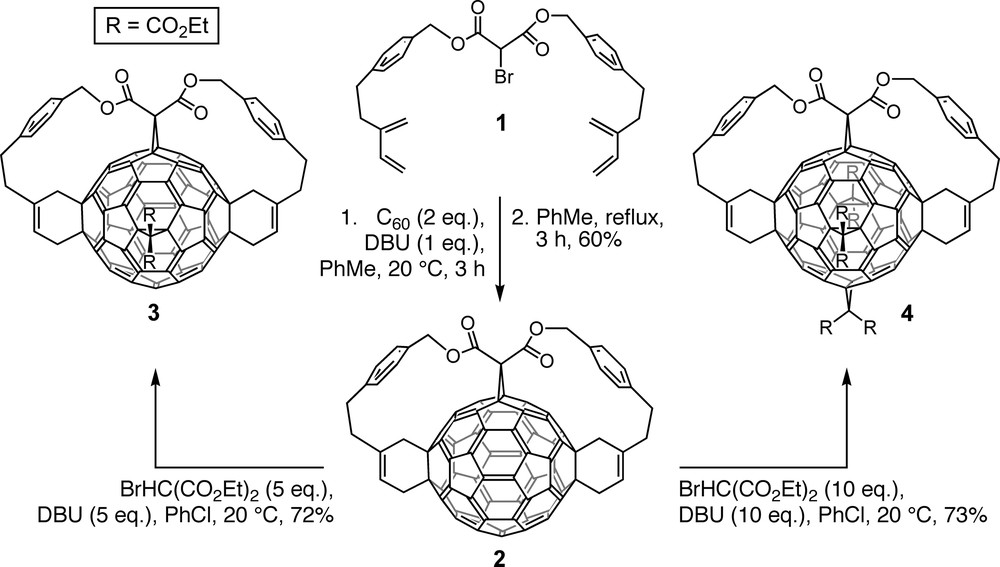
Regioselective formation of e,e,trans-1 trisadduct 2 by tether-directed remote functionalization and selective further transformation into tetrakisadduct 3 and hexakisadduct 4.
Bingel reaction of 2 with five and 10 equivalents of diethyl 2-bromomalonate and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) regioselectively afforded tetrakisadduct 3 and hexakisadduct 4, respectively (Scheme 1) [5,16]. The latter displays a pseudo-octahedral addition pattern and its residual π-chromophore corresponds to a cubic cyclophane with eight benzenoid rings that are located at the corners of a cube and interconnected along its edges [5,16,18,19]. The generation of pentakisadducts by bromomalonate addition to tetrakisadduct 3 afforded unequal amounts of two regioisomers in which the fifth addend occupies either of the two remaining positions that become functionalized en route to hexakisadduct 4 [5,16,18–20]. In the more abundantly formed pentakisadduct, the bond functionalized last (e in relation to the original anchor) is positioned doubly eface and singly eedge relative to the neighboring addends; reverse relationships are found for the minor pentakisadduct resulting from addition of the fifth group trans-1 to the anchor. It can be inferred that double bonds in eface position relative to an existing addend are more reactive than eedge bonds, a finding supported also computationally [21] and by other experimental work [22,23].
Introduction of a diethynylmethano residue trans-1 to the anchor of a slightly modified pentakisadduct and treatment of the resulting hexakisadduct under Eglinton–Glaser conditions afforded tri- and tetrameric macrocycles 5 and 6 (Fig. 2). They represent solubilized derivatives of fullerene-acetylene hybrid carbon allotropes with the general formula C65(n + 2) [16,18]. Their MALDI-TOF (matrix-assisted laser-desorption time-of-flight) mass spectra showed a sequential loss of fullerene units, leading to the ions of mono-fullerene adducts of cyclo[15]- and cyclo[20]carbon.
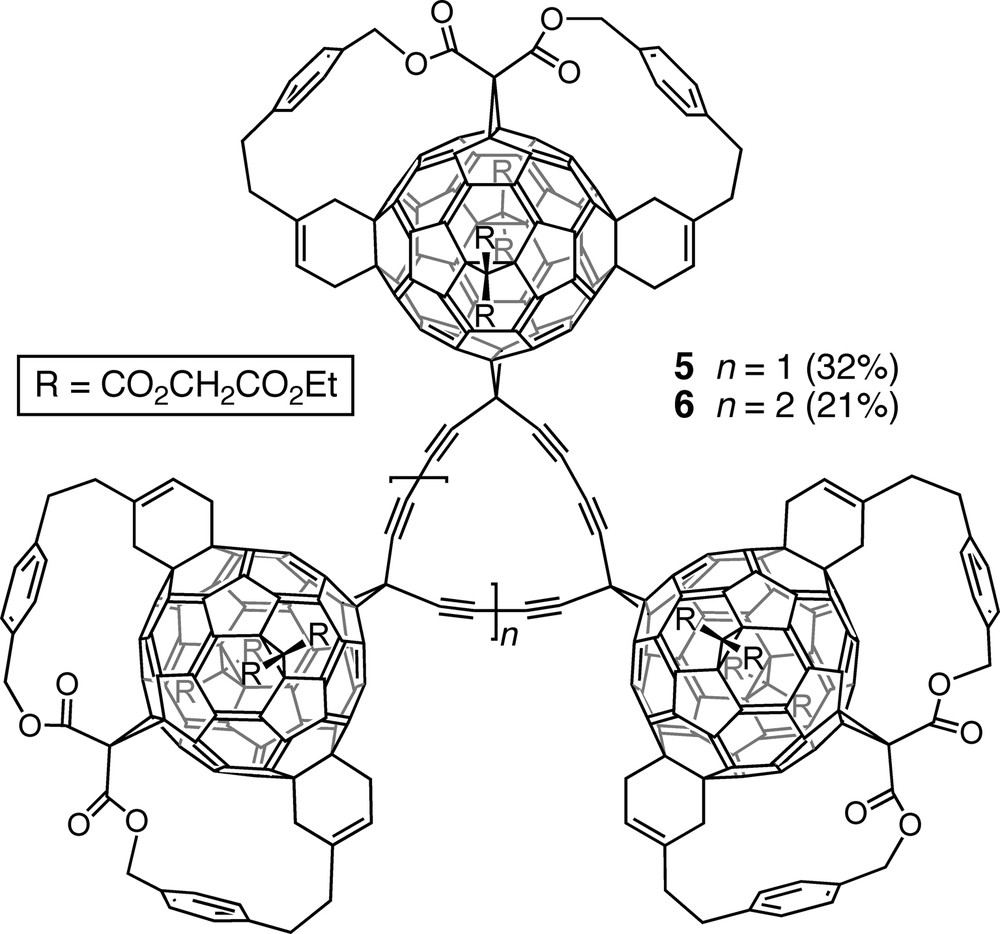
Solubilized derivatives of fullerene-acetylene hybrid carbon allotropes C195 and C260, obtained by Eglinton–Glaser coupling of the corresponding diethynylmethanofullerene precursors.
A truly templated method requires the final removal of directing auxiliaries employed in the course of a synthesis. To eliminate the tethers from 4 (Scheme 1), direct retro-Diels–Alder reaction was not feasible because of the high thermal stability of fullerene-fused cyclohexene rings [24]. However, following a multistep procedure by An et al. [25], transformation of the fullerene-fused cyclohexene to cyclohexadiene units (7, Scheme 2) and in situ treatment of the latter with dimethyl acetylenedicarboxylate afforded, in a Diels–Alder/retro-Diels–Alder sequence, tetrakisadduct 8 which was finally transesterified to octakis(ethyl ester) 9 [26,27]. As confirmed by X-ray crystallography [27], 9 includes two 6–6 bonds in all-e position relative to the addends in place which can be exploited for further molecular scaffolding [28,29]. Tetrakisadduct 9 was obtained independently by Kräutler and co-workers, as reported by Schwenninger et al. [22] and Kräutler et al. [30], through a completely different template-directed approach including, as a key step, a topochemically controlled, regioselective anthracene transfer in heated crystals of the monoadduct resulting from Diels–Alder addition of anthracene to C60 [22,30].
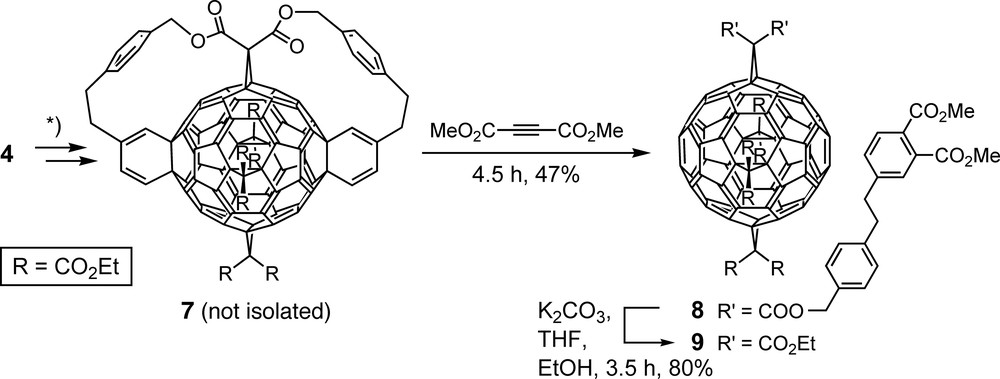
Removal of the cyclohexene units and the tethers of hexakisadduct 4 (Scheme 1) under generation of tetrakisadduct 9 with all addends located on an equator of the carbon spheroid. *) Synthetic details: see [25–27].
Derivatives with an added or inserted methylene group were obtained from pyrazolofullerene 10, product of the [3 + 2] cycloaddition between the according pentakisadduct precursor and diazomethane [31]. Whereas thermal extrusion of dinitrogen afforded hexakisadduct (±)-11, photolysis of 10 led to a 1:1 mixture of homofullerene (±)-11 and methanofullerene 12 (Scheme 3). On the basis of the analogy to the products of N2-elimination from diazoalkane–toluene adducts [32] as well as high-level calculations [33], thermolysis was proposed to pass through a Möbius aromatic transition state in an eight-electron orbital-symmetry-controlled [2πs + 2πs + 2σs + 2σa] process [31,33]. It leads to an intermediate norcaradiene substructure that cycloreverts to the cycloheptatriene unit of homofullerene derivative (±)-11. In contrast, photolysis of 10 presumably proceeds through a diradical mechanism, resulting in a 1:1 mixture of (±)-11 and 12. Further addition of diazomethane provided di- and trihomofullerene derivatives in which all inserted CH2 groups depart from the same central double bond. However, complete isolation of the latter from the residual fullerene chromophore by introduction of a fourth methylene group failed, presumably for steric reasons.
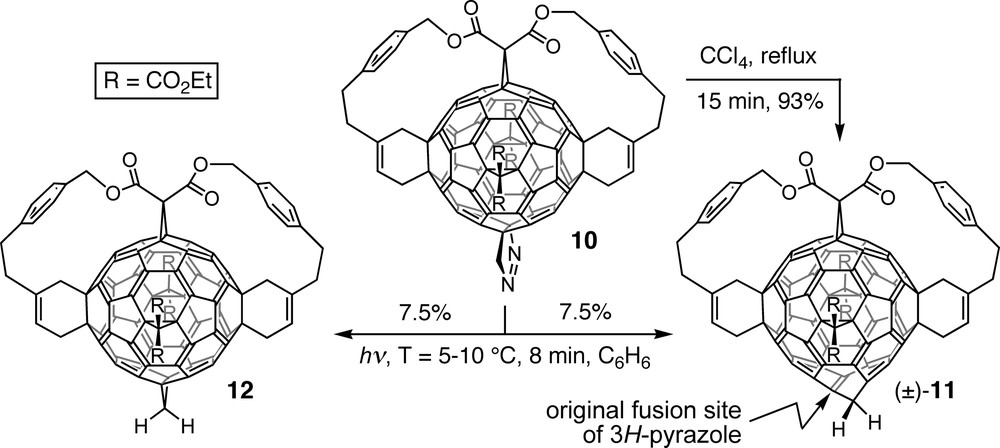
Thermal and photochemical extrusion of dinitrogen from pyrazolofullerene 10, affording homofullerene (±)-11 and a mixture of the latter and methanofullerene 12, respectively.
In most subsequent applications of the tether-directed remote functionalization of fullerenes, the formal distinction between anchor and reactive group did no longer make sense. In fact, a practical simplification of the method consists in treating the carbon cage with a reagent in which two or several identical functional groups are interconnected by a tether; in such one-pot multi-stage additions, each addend already attached to the fullerene acts as an anchor for the remaining reactive groups.
4 Crown ether tethers
Among the isomers obtained by addition of monofunctional, independent reagents, trans-1 bisadducts of C60 (Fig. 1) are generally the least abundant for both kinetic and statistical reasons [10,21]. Also, the design of a tether sufficiently large to span the distance between two poles of the carbon sphere and conformationally homogeneous enough to provide good regioselectivity posed a considerable challenge. The dibenzo-[18]crown-6 macrocycle which can act simultaneously as a spacer and an ionophore was found to present an elegant solution to this problem.
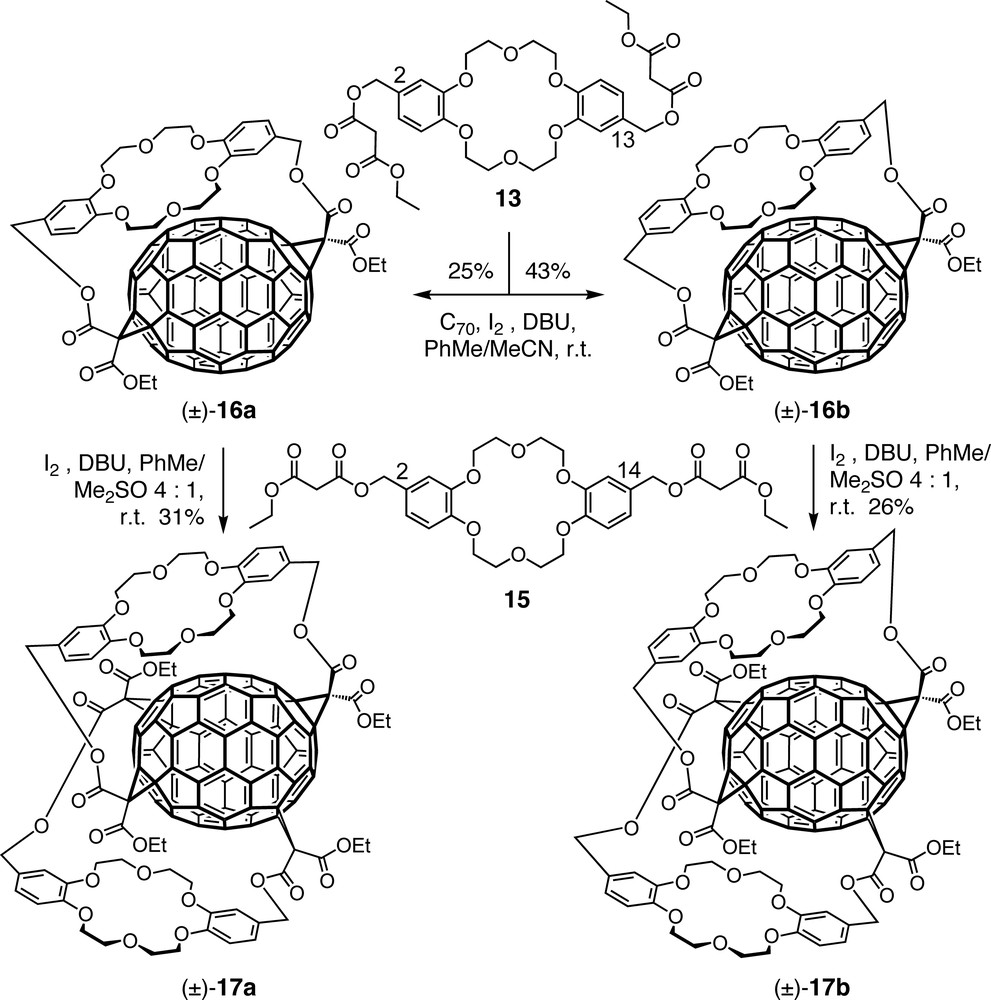
Addition of a bismalonate derived from anti-disubstituted dibenzo [18]crown-6 (13, Scheme 4) to C60 afforded trans-1 bismethanofullerene derivative (±)-14 (Scheme 5) in 30% yield together with 3% of the corresponding trans-2 isomer [34]. Addition of KPF6 to the reaction mixture led to an enhanced yield (50%) and regioselectivity (only trans-1) through an additional template effect originating from the complexation of K+ ions by the ionophore which reduces its conformational flexibility [35]. The complex between (±)-14 and KPF6 was characterized by X-ray crystal-structure analysis which confirmed the close tangential orientation of the ionophore atop the carbon cage. Reaction of C60 with a constitutionally isomeric tether derived from syn-disubstituted dibenzo [18]crown-6 (15, Scheme 4) regioselectively afforded a trans-3 bisadduct.
Single and double addition of dibenzo [18]crown-6-derived bismalonates to C70.
Multistep transformation of trans-1 bis-adduct (±)-18 into hexakisadduct (±)-22 with a double-helical, inherently chiral addition pattern.
Although the trans-1 addition pattern is D2h-symmetric and bismalonate 13 is achiral, the product of the double Bingel addition of 13 to C60 is a racemate ((±)-14, Scheme 5). The chirality of the C2-symmetric bisadduct results from the restricted conformational mobility of the crown ether fixed in close proximity of the fullerene sphere to which it can expose only a single face and, in conjunction with the substitution pattern of the dibenzo-fused heterocycle, constitutes an element of planar chirality. The racemic nature of (±)-14 was demonstrated in 1H-NMR-based complexation studies with enantiomerically pure potassium (S)-1,1′-binaphthalene-2,2′-diyl phosphate and potassium (1S)-camphorsulfonate [35].
Dibenzo [18]crown-6-derived bismalonates were also successfully used in the first tether-directed remote functionalization of C70 [36–38]. Macrocyclization of this fullerene with bismalonate 13 afforded four stereoisomers, namely racemates (±)-16a and (±)-16b, with complete regioselectivity (Scheme 4) [36,38]. The chirality of the [70]fullerene–crown ether conjugates is associated with an inherently chiral fullerene addition pattern in combination with the planar chiral unit of the anti-disubstituted crown ether. Interestingly, the addition pattern of (±)-16a and (±)-16b differs from that of the major product of two independent additions of untethered diethyl malonate units to C70 [39]. A sizeable template effect was noted when the synthesis of (±)-16a and (±)-16b was carried out in the presence of KPF6: whereas the regioselectivity remained unaffected, the diastereoselectivity increased and the total yield went up from 41% to 68%.
When bisadducts (±)-16a and (±)-16b were allowed to separately react with syn-disubstituted dibenzo[18]-crown-6 derivative 15, tetrakisadducts (±)-17a and (±)-17b, respectively, with C70 sandwiched between two crown ethers, were obtained regio- and stereoselectively [37,38]. It should be pointed out that the syn substitution pattern of 15 avoids introduction of another planar chiral unit.
Addition of KPF6 to solutions of fullerene-crown ether conjugates (±)-14 (Scheme 5), (±)-16a, (±)-16b, (±)-17a, or (±)-17b (Scheme 4) led to an anodic shift of the first reduction potential of the fullerene moiety, as shown by CV (cyclovoltammetry). These first-observed cases of cation-complexation-induced alterations of the redox potentials of fullerene derivatives are due to an electrostatic effect of the K+ ion fixed in close proximity of the carbon spheroid. The measured shift is particularly large for (±)-17a and (±)-17b (+170 mV, 0/–1 redox couple) in which cases the fullerene is sandwiched between two cations [37,38]. Incorporation of C60 derivative (±)-14 in the membrane of an ion-selective electrode showed that K+ forms the strongest complex among the alkali metal ions [34,35].
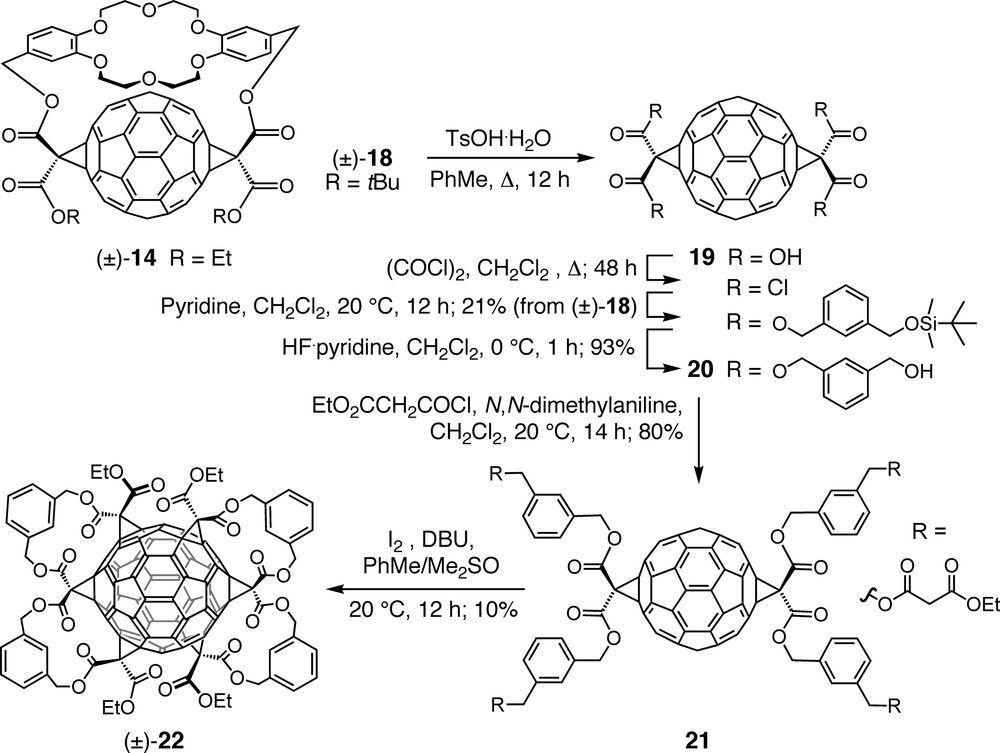
Conjugate (±)-18, a di-tert-butyl ester analog of (±)-14 (Scheme 5), was prepared as a key intermediate en route to a hexakisadduct of C60 with an inherently chiral addition pattern. Acid-catalyzed cleavage of the tert-butyl ester groups and the crown ether template afforded the corresponding tetracarboxylic acid 19 which was transformed over several steps into tetrol 20 and further into tetrakismalonate 21. The final step of the sequence, affording D2-symmetric hexakisadduct (±)-22 as sole product [29,40], consisted of two double tether-directed remote Bingel additions, multiple macrocyclizations which had previously been described as ‘clipping reactions’ and given access to tris- and tetrakisadducts of C60 [41]. X-ray crystallography of (±)-22 confirmed all six cyclopropane rings to be arranged in a double-helical fashion with the corresponding axis passing through the unfunctionalized poles of the carbon sphere. π-Electron conjugation is maintained between the two hemispheres of shiny red (±)-22 via two (E)-stilbene-like bridges. This constitution leads to rather different chemical reactivity and physical properties in comparison to the pale yellow hexakisadducts with a pseudo-octahedral addition pattern (e.g., 4, Scheme 1). Hexacyclopropafullerene (±)-22 readily undergoes further Bingel additions at the two sterically most accessible polar 6–6 bonds under formation of orange-yellow heptakis- and octakisadducts [29,40].
5 Stereoselective synthesis of multiadducts of C60
5.1 Stereoselective targeting of cis addition patterns
The C2-symmetric addition patterns cis-2, trans-2, and trans-3 (Fig. 1) are distinguished by their inherent chirality [11–13], and their stereoselective generation by use of chiral, enantiopure tethers is of high interest. Our first attempts towards this goal were based on spacers derived from easily available enantiopure threitol, which, in the meantime, has proven to be a versatile chiral auxiliary for the stereoselective multifunctionalization of fullerenes [8,13].
Bingel reaction between C60 and either (R,R)-23 or (S,S)-23 afforded the enantiomeric cis-3 bisadducts (R,R,f,sC)-24 and (S,S,f,sA)-24 (Scheme 6), respectively, as minor products besides the corresponding cis-2 (Fig. 1) bisadducts (R,R)-25 and (S,S)-25, the addition pattern of which is achiral [41,42]. Removal of the chiral tethers from (R,R,f,sC)-24 and (S,S,f,sA)-24 by transesterification with ethanol yielded tetraethyl esters (f,sC)-26 and (f,sA)-26, respectively, as pure enantiomers exhibiting strong CD (circular dichroism) bands between 250 and 750 nm with Δε values approaching 150 M–1 cm–1. Such strong Cotton effects are typical for chiral π-chromophores of fullerene derivatives with an inherently chiral addition pattern [11–13]. Absolute configurations could be assigned as [CD(–)488]-(f,sC)-26 and [CD(+)488]-(f,sA)-26 by a comparison between experimental and calculated CD spectra [43].
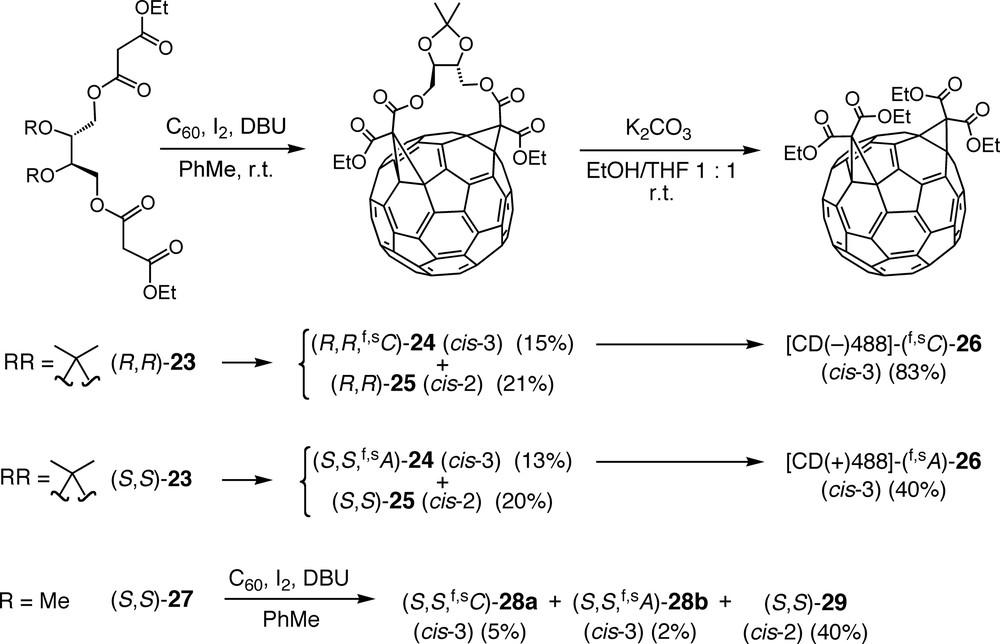
Regio- and diastereoselective addition to C60 of bismalonates derived from enantiopure threitol and cleavage of the tether auxiliary from the cis-3 bisadducts (R,R,f,sC)-24 and (S,S,f,sA)-24, affording (f,sC)-26 and (f,sA)-26, respectively, as pure enantiomers with an inherently chiral addition pattern.
On the other hand, the open-chain l-threitol-derived tether of bis-malonate (S,S)-27 afforded the diastereoisomeric cis-3 bisadducts (S,S, f,sA)-28a and (S,S, f,sC)-28b (Scheme 6) as minor products, next to cis-2 bisadduct (S,S)-29, in a 5:2 ratio which reflects a rather poor diastereoselectivity [44]. It can be concluded that the reduced conformational flexibility of tethers (R,R)-23 and (S,S)-23, associated with their cyclic acetal function, is essential for the quasi-complete diastereoselectivity observed in the formation of (R,R,f,sC)-24 and (S,S,f,sA)-24.
The importance of the conformational homogeneity of the tethers used in multifunctionalizations of fullerenes is also nicely illustrated by the following example: Macrocyclization between enantiopure (R,R)-butane-2,3-diyl [45] bismalonate and C60 afforded cis-3 bisadduct (R,R,f,sA)-30 (Fig. 3) as sole regioisomer in 24% yield and with > 97% de (= 100 × ([(R,R,f,sA)-30] – [(R,R,f,sC)-30])/([(R,R,f,sA)-30] + [(R,R,f,sC)-30])) [46]. According to semiempirical calculations, the substituents of the ethylene tether fragment of (R,R,f,sA)-30 adopt a staggered conformation with gauche relationships between the two methyl as well as between the two ester groups and an antiperiplanar (ap) arrangement of the H-atoms (Newman projection of Fig. 3). Fixation of the ethylene unit of the tether in this conformation by incorporation in a bismalonate derived from trans-cyclohexane-1,2-diol even led to an increased yield (32%) of exclusively and diastereoselectively formed cis-3 regioisomer upon reaction with C60. Conversely, when the glycolic fragment is locked in an unproductive conformation, for example in trans-cyclopentane-1,2-diyl or exo,cis- and exo,trans-norbornane-2,3-diyl bismalonates, Bingel macrocyclization with C60 failed almost completely. The absolute configuration of (R,R,f,sA)-30 was determined by comparing the results of an NMR-spectroscopic and a computational conformational analysis of the tether fragment [46]. The result is in accord with the near-identity of the CD spectra of (R,R,f,sA)-30 and (f,sA)-26 (Scheme 6) which reflects their matching chiral π-chromophores.
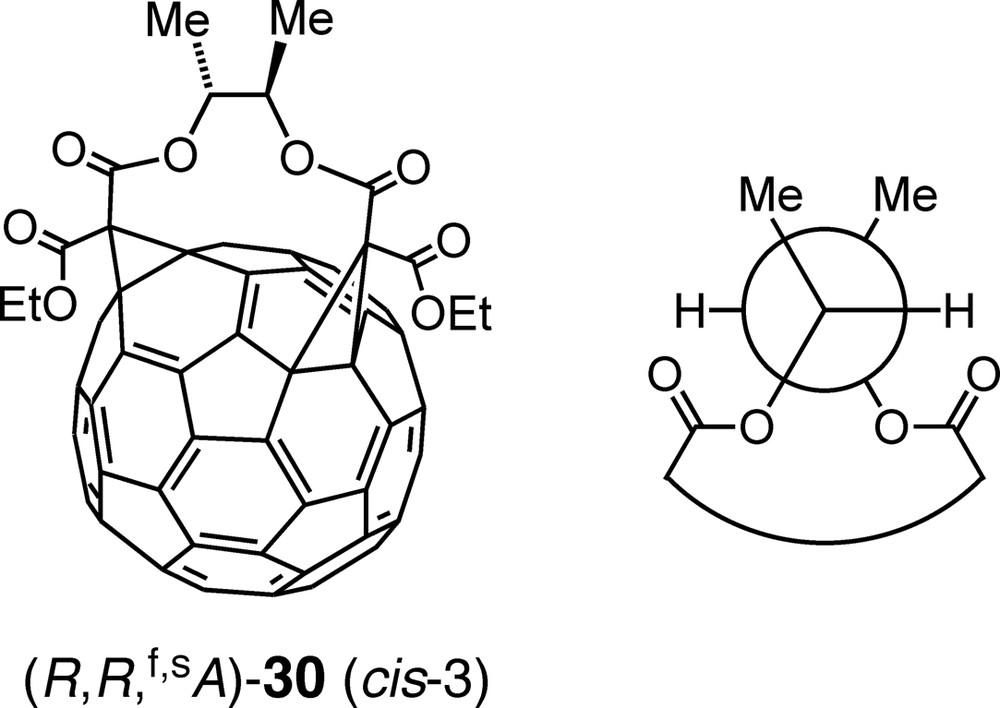
Bisadduct (R,R,f,sA)-30, obtained by diastereoselective addition of (R,R)-butane-2,3-diyl bismalonate to C60 (left) and Newman projection of the preferred conformation of the tether fragment (right).
5.2 Stereoselective targeting of trans addition patterns: tethers derived from various Tröger-type bases
The difficulty of finding tethers spanning the two hemispheres of C60 and simultaneously inducing a high regioselectivity was mentioned above. Regio- and stereoselective targeting of the inherently chiral trans-2 and trans-3 addition patterns seemed all the more challenging since it requires large and conformationally constrained chiral tethers. Derivatives of the C2-symmetric Tröger base were envisaged as suitable for this purpose: they are characterized by rigidity and a folded geometry with nearly orthogonal aromatic rings as distinctive structural features. In addition to the attainment of the inherently chiral trans-2 and trans-3 functionalization patters, such tethers allowed the high-yielding regioselective preparation of trans-1 and trans-4 bisadducts of C60 [47].
Thus, double Bingel cyclopropanation of C60 with bismalonate (±)-31 including a Tröger base assembled from a para-substituted aniline, afforded trans-2 bisadduct (±)-32 as major regioisomer next to a minor amount of e isomer (±)-33 (Scheme 7) [47]. The addition patterns of these regioisomers as well as those of the compounds described below were unequivocally established by UV/Vis and NMR spectroscopies. The absorption spectra of malonate bisadducts of C60 in the region between 400 and 800 nm are mainly determined by the structure of the fullerene chromophore and can, therefore, be used as fingerprints for the corresponding addition patterns [48]. When the above Bingel addition was carried out with enantiomerically pure malonates (S,S)-31 and (R,R)-31, obtained from the racemate by HPLC (high performance liquid chromatography) on a chiral stationary phase, bisadducts (S,S,f,sA)-32 and (R,R,f,sC)-32, respectively, were obtained with complete diastereoselectivity. The absolute configurations of the enantiomers were determined by comparison of their CD spectra with those of previously reported enantiomerically pure derivatives of C60 with the same functionalization pattern [48,49]. Under the assumption that the thermodynamic stability of such bisadducts is reflected in the transition state of the second cyclopropanation, the relative heats of formation of (S,S,f,sA)-32 and its hypothetical diastereoisomer (S,S,f,sC)-32′ were calculated using the semiempirical PM3 method. The computed difference ΔΔH = 29.4 kcal mol–1 in favor of the actually formed (S,S,f,sA)-32 rationalizes the experimental observation very well.
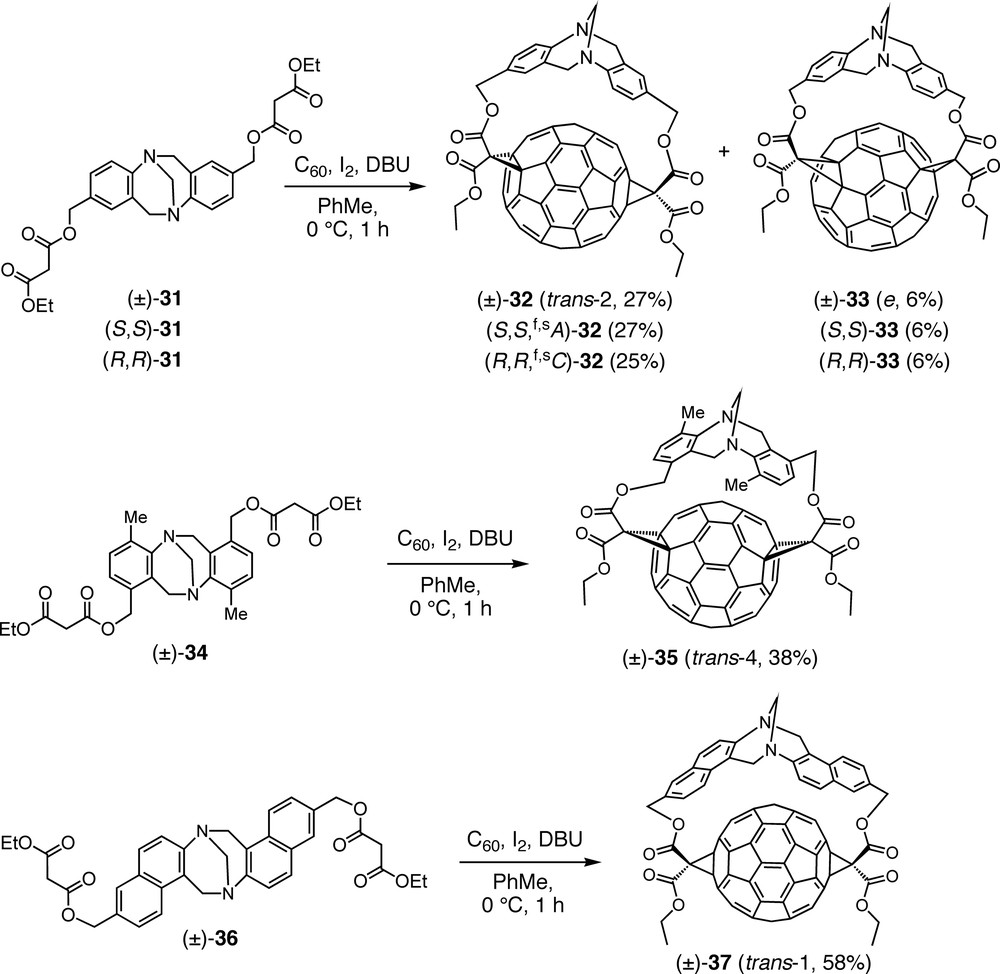
Regio- and stereoselective Bingel macrocyclizations between C60 and racemic or enantiopure bismalonates derived from various Tröger-type bases.
Variation of the geometry of the tether by attachment of the malonate residues to the meta position of the aniline subunits of the Tröger base ((±)-34) afforded exclusively trans-4 regioisomer (±)-35 upon reaction with C60 (Scheme 7) [47].
Alteration of the core of the Tröger base, on the other hand, by switching from an aniline derivative as precursor to a naphthyl-2-amine substituted at C(6) (bismalonate (±)-36) led to completely regioselective and high-yielding formation of trans-1 bisadduct (±)-37 (Scheme 7) [47]. Although crystallization of this compound is tricky, a single crystal suitable for X-ray analysis could be isolated from a solution of (±)-37. Remarkably, it contained only the (R,R)-configured enantiomer and this seems to be the first case of spontaneous resolution of a racemic fullerene derivative.
In an attempt to achieve also the inherently chiral trans-3 functionalization pattern, bismalonate (±)-38, including two 4′-substituted 4-aminobiphenyl units in the Tröger base core, was added to C60. The reaction was completely regioselective, affording only the targeted trans-3 addition pattern, but in the form of four stereoisomers consisting of two pairs of enantiomers, namely (±)-39a and (±)-39b (Scheme 8) [47]. Whereas all the above Tröger base-fullerene conjugates are out,out isomers (out refers to the orientation of the ethoxycarbonyl substituents of the cyclopropane rings), the C1 symmetry of (±)-39a and (±)-39b points at in,out isomers. Separate syntheses with enantiomerically pure starting materials, (S,S)-38 and (R,R)-38, allowed the isolation of all four stereoisomers, i.e. (S,S,f,sA)-39a and (S,S,f,sC)-39b, as well as (R,R,f,sC)-39a and (R,R,f,sA)-39b, respectively. Absolute configurations were assigned by comparison of the CD spectra with the published data of enantiomerically pure C60 derivatives with the same addition pattern [48,49]. Whereas the CD spectra of enantiomers are exact mirror images, those of diastereoisomers show some deviation from this relationship due to the superposition of the Cotton effects originating from the fullerene and the Tröger base chromophores.
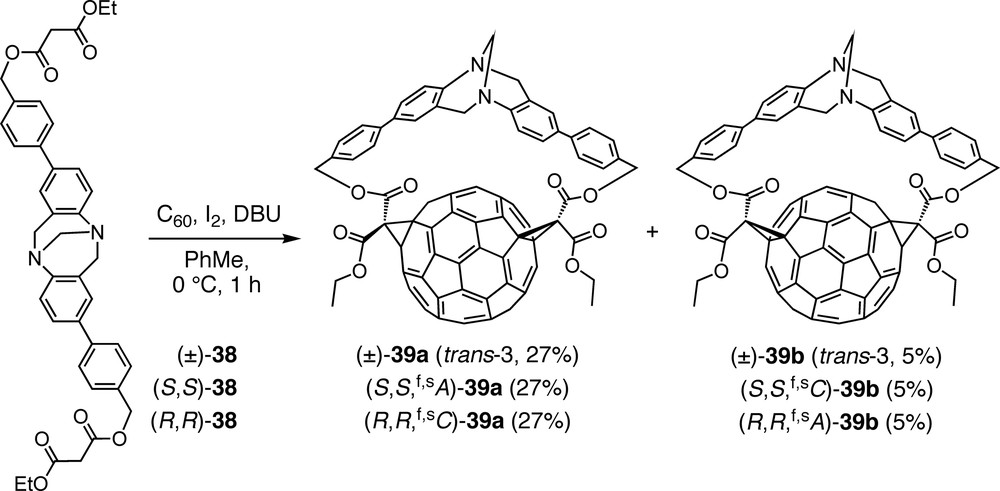
Regio- and stereoselective synthesis of in,out trans-3 bisadducts of C60 using enantiopure or racemic extended Tröger bases including biphenyl units.
These asymmetric syntheses using the structural motif of the Tröger base as a ‘chiral auxiliary’ allowed the first stereoselective targeting of the inherently chiral trans-2 and trans-3 addition patterns of buckminsterfullerene. Tröger-type bases with different angles between the planes of the arene rings as well as representatives derived from thiophene and benzothiophene were successfully used by Ishida et al. [50] in a different study on the regio- and stereocontrolled addition of accordingly tethered bismalonates to C60.
6 Addition of tethered tris- and tetrakismalonates to C60
The first direct threefold Bingel macrocyclization consisted in the addition to C60 of tripodal trismalonate (±)-40 in which three propanedioate moieties are attached to a chiral CTV (cyclotriveratrylene) core (Scheme 9) [51]. The two racemic trisadducts isolated from this reaction were assigned as e,e,e and trans-3,trans-3,trans-3 regioisomers (±)-41 and (±)-42. Both structures present an interesting superposition of an inherently chiral fullerene addition pattern (possible configurations (f,sC) and (f,sA)) and a conformationally chiral CTV unit (possible configurations (P) and (M)) which can lead to a total of four stereoisomers (two pairs of enantiomers) for each regioisomer. Intriguingly, a detailed mathematical analysis revealed that the corresponding topological stereoisomers, besides being topologically chiral, amount to four in the case of the e,e,e adduct but only to two in that of the trans-3,trans-3,trans-3 isomer [52]. This is a consequence of the fact that the median plane including the addends cuts the carbon sphere into unequal fractions in the former ((±)-41), but into equal halves in the latter ((±)-42) structure.
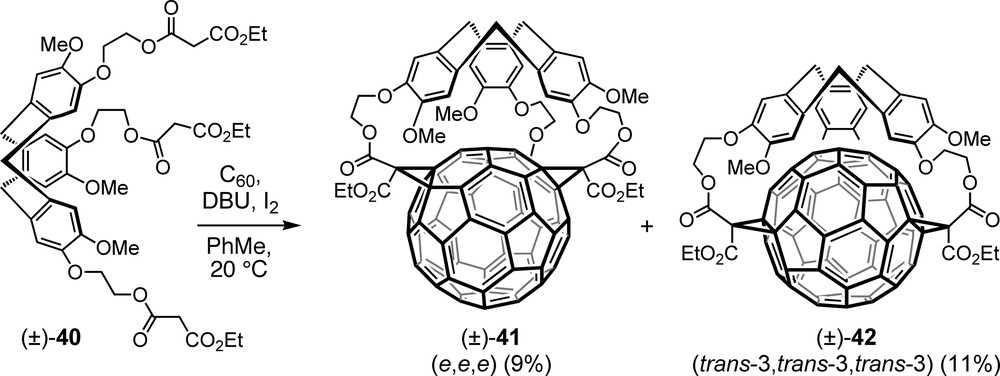
Products of the first one-pot direct addition of a trismalonate to C60.
Another interesting approach towards regioselective bis-, tris-, and tetrakis-functionalization of C60 is based on the Bingel reaction with flexible cyclooligomalonates and was developed by Reuther et al. [53]. Whereas fullerene adducts resulting from addition of oligomalonate macrocycles with identical oligomethylene chains between the carboxy groups are generally characterized by rotational symmetry, those produced from cyclooligomalonates with unequal oligomethylene chains lead to other addition patterns.
7 Further tethers
Due to their easy availability, xylylene tethers have been applied to a large variety of tether-directed multiple functionalizations of C60 by the Diederich group [41,54] and by others [8]. They show different regioselectivities depending mainly on the substitution pattern of the core benzene ring but also on other factors such as the specific functional groups involved. A particularly elaborate example is catenane (±)-43 (Fig. 4), the p-xylylene tether of which induces a trans-4 pattern of biscyclopropanation, thereby becoming an element of planar chirality due to its restricted conformational mobility in combination with its particular substitution pattern [55]. Tethers derived from m-xylene have been extensively used over the past years for the synthesis of a large structural variety of fullerene-derived dendrimers; these appear to be very promising tectons with regard to supramolecular chemistry and materials science applications [56].
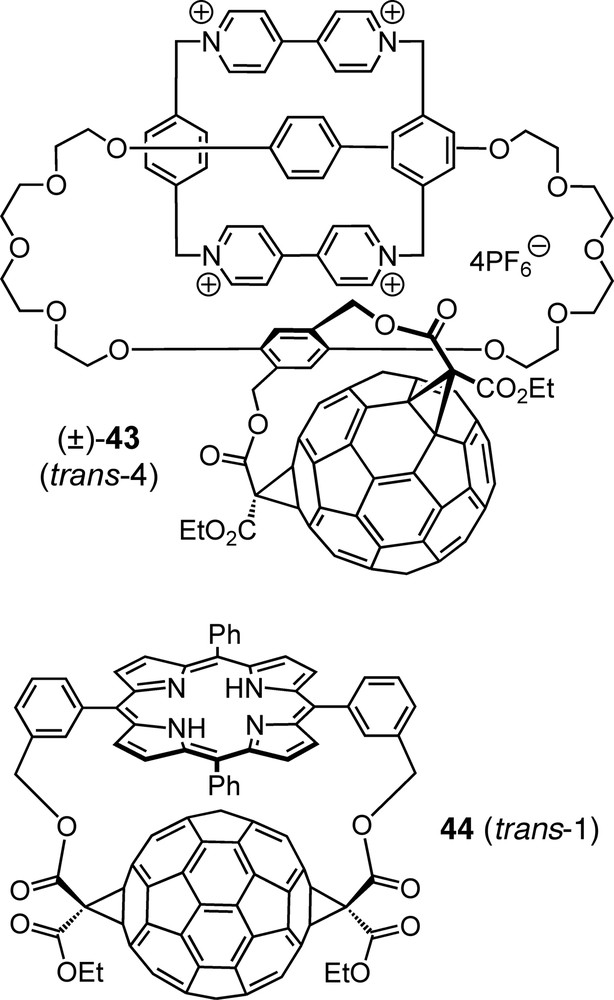
Planar-chiral fullerene-derived catenane (±)-43 and fullerene–porphyrin conjugate (±)-44 — examples of [60]fullerene bisadducts with interesting structure and function, obtained regioselectively by tether-directed remote functionalization.
Fullerene–porphyrin conjugates continue to be one of the hottest research topics in fullerene chemistry due to their potential as model systems mimicking natural photosynthetic reaction centers by converting photonic excitation energy to chemical potential in the form of long-lived charge-separated states [57]. The use of porphyrins as tethers in the synthesis of such dyads, e.g. (±)-44 (Fig. 4), allows not only for regioselective addition to the fullerene but also for precise control of the geometric parameters which have a crucial influence on the photophysical and electron-transfer properties [58]. Interestingly, extension of the chains linking the porphyrin to the malonates by one C-atom on each side leads to exclusive formation of the corresponding trans-2 bisadduct [59].
Further bismalonates used by the Diederich group in Bingel macrocyclizations include tether residues such as trans-but-2-ene-1,4-diyl [42,60], 1,10-phenanthroline-2,9-dimethylene, 1,10-phenanthroline-2,9-bis(1,4-phenylene), and 9,9′-spirobifluorene-2,2′-dimethylene [41]. They afforded fullerene adducts showing interesting structural features with varying regio- and, in the occurrence, stereoselectivities.
8 Conclusion
Roughly a decade after the introduction of the tether-directed remote functionalization into fullerene chemistry [5], this method has made all addition patterns of C60 available with fair to high regioselectivity [7,8]. Among them, the cis-1 pattern is limited to particular cases most of which can be considered as multiple anellations of the fullerene [8]. The reason for this limitation is the steric hindrance between most addends in such an arrangement, which precludes, for example, the formation of Bingel-type cis-1 bisadducts. As to the inherently chiral cis-3, trans-3, and trans-2 functionalization patterns, they are now available not only regio- but also diastereoselectively when enantiopure tethers are used in the macrocyclizations. Broader avenues to regio- and/or stereoselectively formed trans bisadducts have opened up only recently through the introduction of a variety of tethers derived from the Tröger base [47]. If one makes abstraction of the inaccessible cis-1 bisadducts, Bingel-type chemistry has proven to be particularly versatile and adaptable to the various functionalization patterns. As to e bisadducts, tether-directed biscyclopropanation usually generates a new stereogenic center [13], the stereocontrol of which has to be further investigated. Also, the stereoselective generation of non-inherently chiral functionalization patterns calls for more profound exploration. The same accounts for the selective generation of higher adducts of C60 by tether-directed remote functionalization although some important progress in this area has been made during the past few years [51–53].
In the chemistry of C70, the tether-based methodology has provided access to only a few functionalization patterns, albeit with good regio- and stereoselectivities [36–38]. As to the spacer-controlled multifunctionalization of other higher fullerenes, it remains a terra incognita.
Acknowledgements
The authors thank the co-workers whose names appear in the references for their valuable contributions. Continuing support of fullerene research by the Swiss National Science Foundation is gratefully acknowledged.