

1 Introduction
As shown from electrochemical studies, [60]fullerene is easy to reduce and can accept up to six electrons in six well separated one-electron reduction steps, leading at maximum to the hexaanion C606– [1]. This electronegative character, associated with the very specific structure of the C60 core in which each hexagon is described at best as a cyclohexatriene ring rather than a benzene ring, gives [60]fullerene the reactivity of an electron-deficient polyolefin. As a consequence, functionalizing the C60 core has been carried out in most cases through cycloaddition reactions and nucleophilic additions [2]. In this context, two processes, namely the Prato reaction ([3 + 2] cycloaddition of azomethine ylides) [3] and the Bingel [4] reaction (cyclopropanation of C60 with a dialkyl bromo(iodo)malonate in the presence of a strong base) are particularly used. They allow to obtain a wide variety of compounds in two different families, respectively, fulleropyrrolidines and methanofullerenes, under simple experimental conditions and in good yields.
Electrophilic reagents, of course, are not expected to easily react with the C60 core because of its electronegative character. However, using such reagents might be considered if the starting fullerene moiety is in C60n– anionic form, these anions being likely to react as potential nucleophiles. This strategy may open up new attractive ways for functionalizing [60]fullerene, and increase to some extent the field of [60]fullerene organo-derivatives. In this case, some conditions are expected to be fulfilled in order to make this approach as fruitful as possible: (i) a specific anion C60n–, possessing a defined charge, should be easily obtained, and readily used in subsequent reactions; (ii) the electrophilic moieties prone to react with C60n– should allow to obtain new [60]fullerene derivatives very difficult, or even impossible, to be prepared through more usual methods starting from C60 itself.
As part of this research, we have studied the reaction of chemically generated C602– anion towards various halogenated organic compounds. We have thus prepared new [60]fullerene derivatives, some of them being able to act as building blocks in C60 chemistry and to give rise to novel compounds in the field of organic materials. These results are summarized in this account.
2 Formation of C602– anion
Kadish et al. have first shown that C602– anion can react with saturated iodo or bromo derivatives RI or RBr, giving rise to corresponding bis-adducts R2C60 [5,6]. Interestingly, these results established that new [60]fullerene organo-derivatives could be obtained from C602–. However the electrochemical formation of this ion, as reported in these studies, appeared to be a somewhat limiting factor for a development of this methodology on a preparative scale.
A forward step was accomplished when the formation of dianion C602– was observed to be performed through chemical reduction of C60 upon reaction with an alcanethiolate anion RS–, this ion being then formed in situ from a mixture of an alcanethiol RSH and K2CO3 in acetonitrile under nitrogen atmosphere [7]. The reaction is slow, due to the weak solubility of the carbonate salt in the reaction mixture which limits to some extent the formation of the RS– K+ salt. Besides, J. Chen et al. have obtained C60n– (n = 1–3) salts in the series AnC60(THF)m after reduction of C60 upon reaction of corresponding stoichiometric amounts of alkali metal A (K, Na) with 1-methylnaphthalene in THF at room temperature [8].
We have found a more simple and efficient process, which also avoids using a very ill-smelling alcanethiol. In this case the reducing agent is the commercially available sodium methanethiolate CH3S– Na+. We observed that this latter salt easily reduces [60]fullerene in acetonitrile at room temperature, and gives rise to the sole C602– anion within a few hours (Scheme 1) under inert atmosphere. C602– anion is unambiguously identified from near-IR spectroscopic studies of the acetonitrile medium where it is perfectly soluble, giving an intense dark red color to this solution [9]:
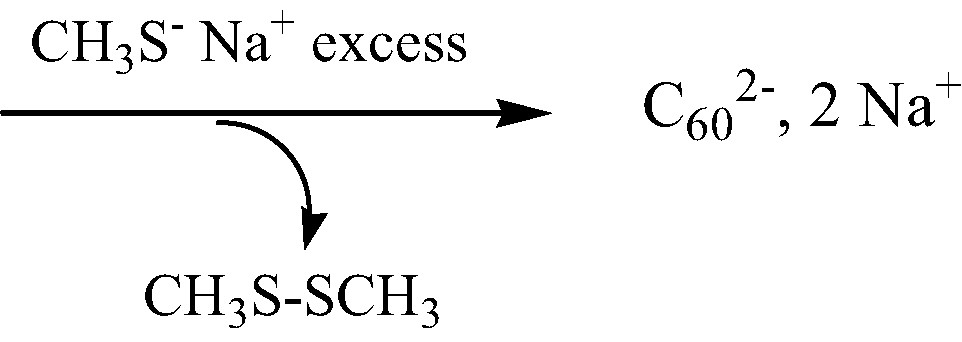
Another chemical process has been reported, in which C602– anion is produced in benzonitrile medium by treating dihydrofullerene C60H2 with tetrabutylammonium hydroxide [10].
3 Reaction of C602– anion with functional halo-derivatives
3.1 Formation of symmetrical bis-adducts R2C60
A great variety of saturated mono-halo derivatives RX has been reacted with C602– in acetonitrile, at room temperature, usually for 24 h.
Some general features characterize the observed results from these halo derivatives [9,11]. The overall yield of the addition, versus the starting amount of C60, lies in the 10–70% range, the lowest yields being generally observed when R is a long chain, while the reactivity of the halo-derivatives decreases in the order RI > RBr > RCl, the chloro-derivatives being able to react only in the case of strongly activated structures, such as benzyl chloride [12].
On the other hand, a mixture of two regio-adducts R2C60 is generally formed, namely the 1,2- and 1,4-isomers 1 and 2 (Scheme 2). The regioselectivity of this reaction is high, the relative molar ratio 1/2 being ca. 9/1 when R = CH3 and 1/9 in other cases (Scheme 2).
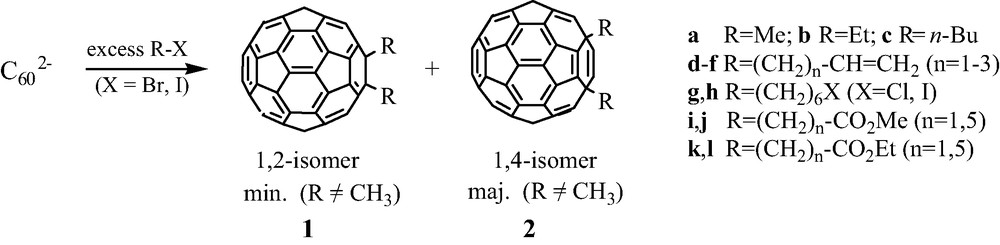
The regiochemistry of this reaction is accounted for by the steric hindrance due to both R groups in the 1,2-positions when R ≠ CH3, whereas theoretical calculations predict a better reactivity of the 2-position compared to the 4-one of the intermediate [RC60]– anion which is first formed whatever the nature of the R group.
The reaction with haloacetates X–CH2–CO2R1 (X = Br, I; R′ = Me, Et) gives rise to particular results [13]. Whereas it is generally quite difficult to separate both 1,2- and 1,4-adducts being formed, major 1,4-(CH2CO2R1)2C60 2i and 2k derivatives are easily isolated as pure compounds through column chromatography. We also observed that these bis-esters can be reduced in good yield (50%) into corresponding bis-aldehydes 1,4-(CH2CHO)2C60 3 through reduction by DIBAL-H at –80 °C. However, a further reduction to alcohols 1,4-(CH2CH2OH)2C60 4 failed, no definite compound being obtained in this case (Scheme 3).
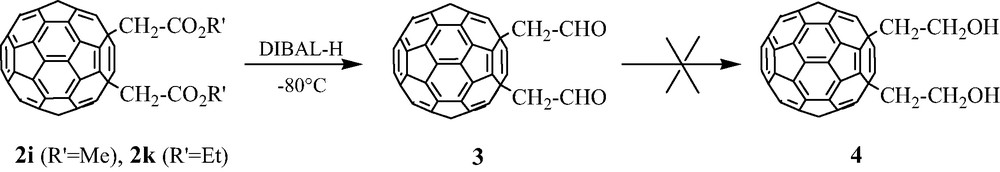
On the other hand, unexpected results have been observed about the electron affinity of the C60 core in these bis-esters 2i and 2k. Whereas C60-derivatives are usually known to be reduced at more negative potentials (average ΔE ~ –100 mV) than C60 itself [14], these 1,4-adducts are characterized by a first reduction potential which remains very close to the one observed for pristine C60.
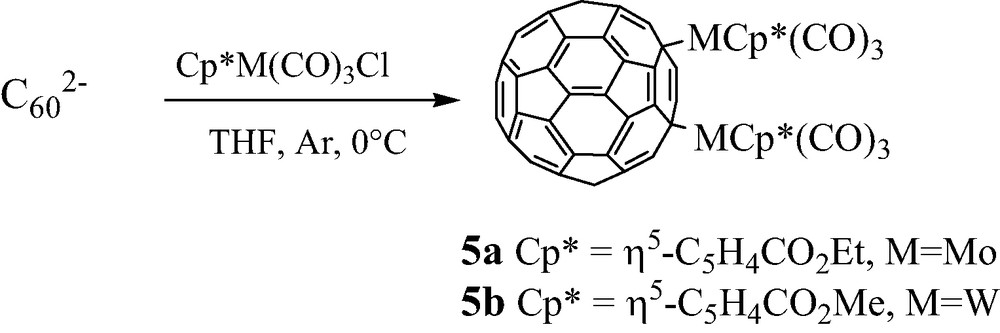
This methodology of functionalization has also been extended to the reaction of organometallic halides with C602– issued from reduction of C60 by potassium/1-methylnaphtalene in THF. Thus, Zhu et al. [15] have obtained two σ-bonded [60]fullerene transition metal derivatives, respectively (μ-C60)Mo2(η5-C5H4CO2Et)2 5a and (μ-C60)W2(η5-C5H4CO2Me)2 5b. These compounds are identified as 1,4-adducts (Scheme 4).
3.2 Formation of cycloadducts
Diiodoalkanes I–(CH2)n–I constitute an important class of reagents when n = 3 or 4, since they lead to cycloadducts 5 or 6 in very good yields (~80%). These results prompted us to extend this reaction to functionalized 1,3-dihalopropane derivatives in order to obtain in one step new cyclopentanofused [60]fullerene able to act as attractive building blocks in further syntheses of multifunctional C60-based covalent assemblies [9]. Thus, the reaction of 1,3-dibromopropan-2-one easily gave rise to the ketone 7 (20% yield) (Scheme 5) [11].
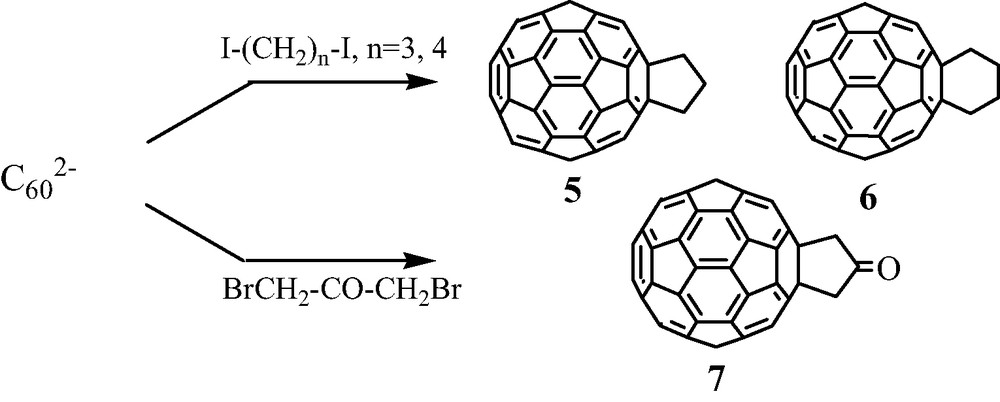
Furthermore, we observed that ketone 7 was easily reduced at room temperature by DIBAl-H into the corresponding alcohol 8 (50% yield) (Scheme 6) [11]. As will be indicated later, alcohol 8 was proven to be a fruitful building block for the synthesis of novel [60]fullerene-based organic materials.

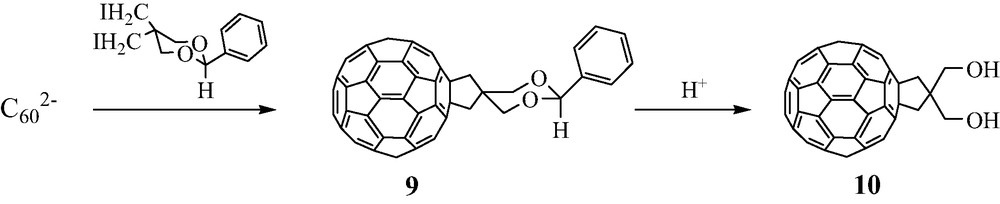
These results led us to search for the synthesis of the diol 10, a relatively few examples of C60 derivatives bearing two alcohol groups being known. From our previous results, the simplest pathway seemed to allow to react C602– anion with the 1,3-diiodopropan-2-ol ICH2–CHOH–CH2I. However, no definite compound could be isolated from this latter reaction. The diol 10 was finally obtained in good yields in two steps (~50% in each step) starting from the diiodo acetal 9 derived from pentaerythritol [16] (Scheme 7).
3.3 Formation of functional mixed bis-adducts RR′C60 (R ≠ R′)
The easy formation of various symmetrical bis-adducts R2C60 starting from C602– anion prompted us to check if the same methodology could be extended to the preparation of functional mixed bis-adducts RR′C60.
Previously, only one C60 adduct bearing two different alkyl groups (t-C4H9)(C6H5–CH2)C60, was reported to be obtained from C602– anion [17]. In the same work, the authors have studied in detail the reaction kinetics of C602– anion with various halo-derivatives RX, from which they have proposed the overall following mechanism in three successive steps (Scheme 8).
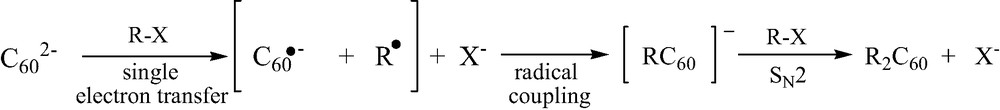
On this basis, the reaction of C602– with t-C4H9I is considered to stop when the [(t-C4H9)C60]– anion is formed, the size of this t-butyl group preventing further fixation of a second one on the C60 core, even in a 4-position. In this case, the introduction of benzyl bromide in the reaction medium allows to add the less bulky benzyl group in the third step, giving rise to (t-C4H9)(C6H5–CH2)C60.
From the above mechanism, it appears that the RX derivative is involved in two quite different steps, a single electron transfer and a SN2 nucleophilic substitution. It thus appeared to be likely to take advantage of these mechanistic differences if we were able to stop the overall process after the radical coupling, irrespective of any steric effect as shown by the t-butyl group.
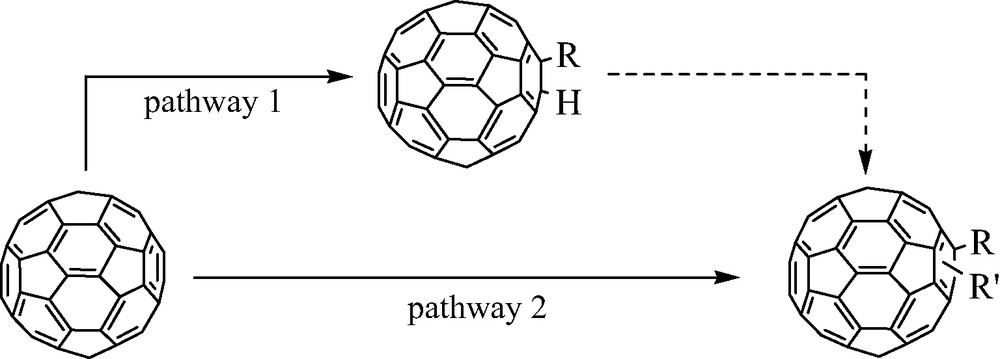
Two possibilities can be considered:
- ● if the RX compound is very reactive, the two first steps will be very rapid. At this stage, the process may be stopped upon addition of a strong acid in order to form then isolate a R(H)C60 derivative (pathway 1). The final RR′C60 will be obtained in a second stage;
- ● if the RX compound is weakly reactive, the two first steps will be slow, and the third one still slower. In this case the RR′C60 final derivative may be obtained in a “one pot” overall process, provided a second R′X compound, much more reactive than RX, is added after the radical coupling (pathway 2) (Scheme 9).
3.3.1 Formation of R(H)C60 compounds
Very reactive halo-derivatives were chosen in the alkyl halo-acetates series, XCH2–CO2R1 11. Following the progress in the reaction with C602– is easy, due to the color change of the solution from red associated to C602– to dark emerald-green characteristic of a [RC60]– anion [18]. As expected, the formation of the [(R1O2C–CH2)C60]– anion is very rapid, and a further addition of an excess of CF3CO2H leads instantaneously to the corresponding dihydrofullerene 1,2-(R1O2C–CH2)(H)C60 12 (Scheme 10).
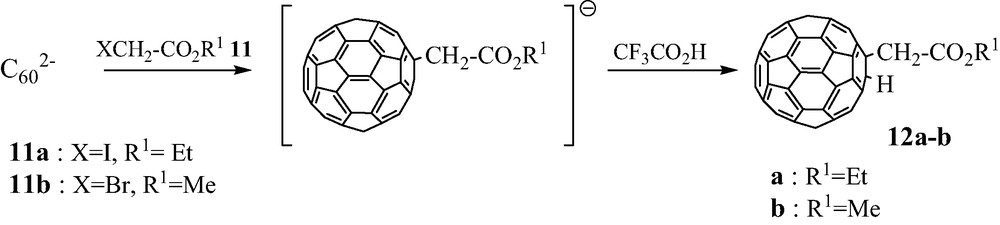
We observed that RHC60 12a–b were formed in 35% yield when CF3COOH is added after 2–3 min reaction at room temperature between C602– anion and corresponding XCH2CO2R1 11 (100 mg C60, 75 ml acetonitrile, 20 eq. RX) [19]. By comparison, under strictly identical experimental conditions, the bis-adducts (CH2CO2R1)2C60 2i–k were obtained in 15–20% yield after 24-h reaction.
A comparable procedure was reported by Meier et al., C602– anion being generated in this case by treating 1,2-dihydro[60]fullerene C60H2 by tetrabutylammonium hydroxide. Various alkyl bromides RBr were used (R=Ph2CH, PhCH2, HC≡C–CH2, CH2=CH–CH2), and gave rise to the corresponding R(H)C60 derivatives in fair to good yields (21% from propargyl bromide, 60% from diphenylmethyl bromide) after an immediate quenching of the reaction mixture by acetic acid [10].
3.3.2 Formation of RR′C60 compounds
A simple process consists of using functional 1,2-(R)(H)C60 12 previously obtained. These compounds are first transformed into their anions [RC60]– upon reaction with tBuOK, then a further reaction of these anions with a second halo derivative R′X allows to easily obtain the expected RR′C60 derivative (Scheme 11).
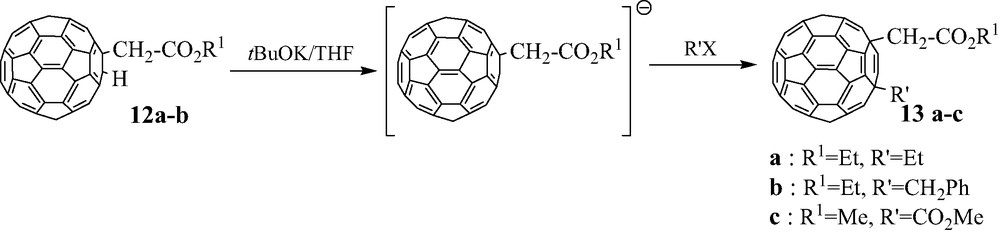
Derivatives 13a–c are obtained in 50–60% yield. Furthermore, it has to be mentioned that the regiochemistry of the second addition is very selective, since in these three adducts both CH2CO2R1 and R′ groups are only in the 1,4-positions on the C60 core [19].
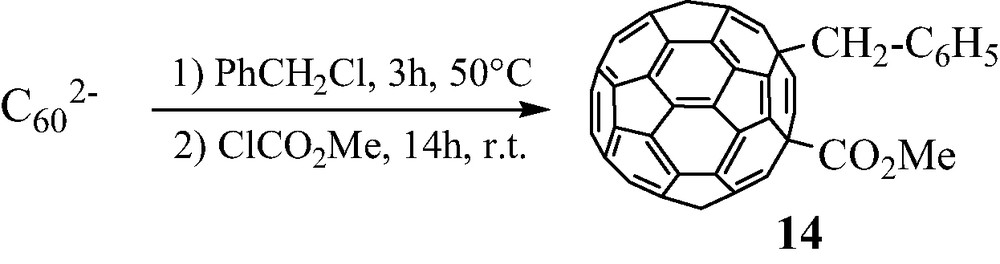
As already indicated, functional mixed RR′C60 compounds might also be obtained according to another strategy, if the first RX derivative is weakly reactive. In this case, the nature of the halogen may be considered. Since the first step of the whole process is a single electron transfer associated with a departure of X– anion, its rate is expected to be slower with X = Cl and faster with X = I. A good candidate is benzyl chloride C6H5CH2Cl, which was already shown to be a weak reagent towards C602– anion in THF [12]. Thus, we observed in a test experiment that the [(C6H5CH2)C60]– anion is produced in acetonitrile between C602– anion and benzyl chloride upon heating at 50 °C for 3 h. A further addition of excess methyl chloroformate ClCO2Me, a priori more reactive than benzyl chloride, allowed to obtain the pure 1,4-(CH2–C6H5)(CO2Me)C60 14 derivative in 15% yield [19] (Scheme 12).
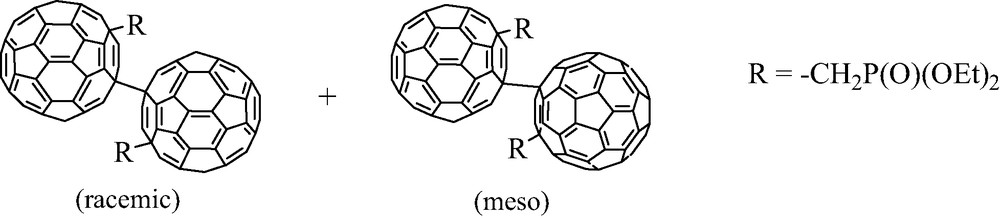
Another particular result was observed by Komatsu’s group. After reaction of C602– anion with diethyl iodomethylphosphonate ICH2P(O)(OEt)2, the [C60(CH2P(O)(OEt)2]– anion does not add a second CH2P(O)(OEt)2 group at room temperature because of its bulkiness, but can react with I2, thus leading to a singly bonded C60 dimer as a mixture of two inseparable isomers [20] (Scheme 13).
4 Optoelectronic properties
Due to their good reduction ability (see Table 1), the bis-esters 1,4-(CH2–CO2R1)C60 2i and 2k appeared as potential candidates as acceptor partners in organic solar cells. In this purpose, the photovoltaic characteristics of different donor–acceptor mixtures have been studied, the donor being poly[2-methoxy-5-(2′ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV). The results show that, for the same w/w donor–acceptor ratio (1/2), the energy conversion efficiency η associated to compounds 2i (0.27%) and 2k (0.4%) is slightly lower, although in the same order of magnitude, than when the acceptor is PCBM (0.7%), the weaker solubility of the bis-esters 2i and 2k in 1,2-dichlorobenzene (ODCB) preventing to work with the more usual w/w donor–acceptor ratio 1/5 [21].
Voltammetric data [E1/2 (V) vs. Fc+/Fc, o-DCB, Pt, 100 mV s−1]
| Compound | E1/2 red1 | E1/2 red2 | E1/2 red3 |
| [60]Fullerene | –1.11 | –1.49 | –1.92 |
| Bis-ester 2i | –1.13 | –1.58 | –2.03 |
| Bis-ester 2k | –1.14 | –1.58 | –2.03 |
From alcohol 8, two C60-TTF-based diads 21a–b were synthesized. Their third-order non-linear optical susceptibilities have been measured in conventional solution (THF) [22] and in nematic liquid crystal [23] (Scheme 14).
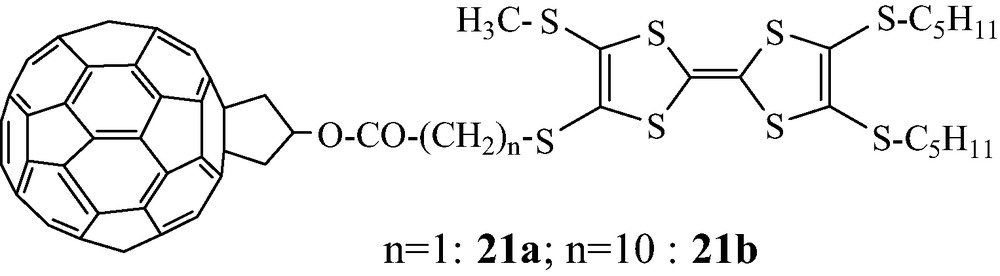
The photophysical properties of 21a–b have also been investigated, and radical ion pairs C60●–/TTF●+ have been characterized from photoinduced electron-transfer processes. The longer lifetime of the ion-pair is associated to the longer spacer between C60 and TTF moieties (N = 10) [24].
More recently, starting from diol 10 we have prepared the triad 22, which includes one C60 unit and two TTF moieties. The study of its photophysical properties indicates that the photoinduced charge separation is likely to take place via the excited triplet state of the C60 moiety, giving rise to a longer lifetime of the radical ion-pair state in non-polar solvents [25] (Scheme 15).
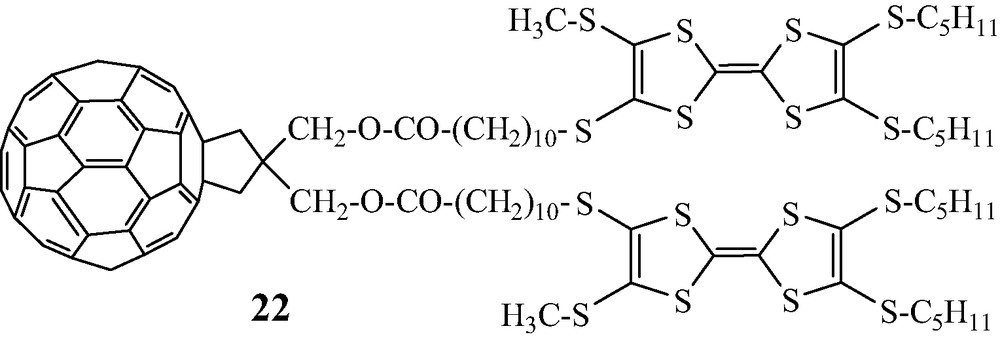
In these C60-TTF-based diads 21a–b and triad 22 the electrochemical HOMO–LUMO gap follows the sequence 22 (1.17 eV) < 21b (1.21 eV) < 21a (1.25 eV).
5 Conclusion
The reactivity of C602– anion has been studied towards a great number of halogenated compounds. The [60]fullerene derivatives issued from these reactions evidenced the interest of this methodology. In particular functional halo-derivatives, thanks to the numerous structures they can offer, greatly enlarge the range of derivatization of the C60 core which can be performed in a few steps. Among these results, we established that a judicious choice of the halo-derivatives and the experimental conditions allows to obtain various functional mixed dihydrofullerenes RR′C60. We have also shown that some of these novel [60]fullerene derivatives are promising building blocks in C60 chemistry, which open up the way to new covalent ensembles in the field of organic materials.
Acknowledgements
The French Ministry of Research and the CNRS are gratefully acknowledged for their support to this work. The author is also deeply grateful to co-workers and collaborators cited in the references for their outstanding contributions in these studies.