

1 Introduction
Following the discovery of [60]fullerene, the chemistry of this fascinating spherical molecule has been intensively investigated. It is now well established that the chemical reactivity of [60]fullerene is typical of an electron-deficient olefin. Indeed [60]fullerene reacts readily with nucleophiles and is a reactive 2π component in cycloadditions. Our group has been involved in the development of various tools for the chemical modification of C60. Over the years, we have shown that fullerene chemistry is governed largely by a combination of steric effects, electron withdrawal by the cages, a drive to increase their aromaticity, and an increase in localization of π-electrons following a first addition. A number of these features are summarized in the present account.
2 Factors governing addition
Fullerenes are not fully aromatic, since the required delocalisation would increase the double bond character of bonds in pentagonal rings, with consequent increase in strain [1]. Thus the double bonds are localised as shown in Fig. 1a, and 1,2-addition occurs readily across a bond between two hexagons (‘6,6-bond’).

The three possible arrangements of two pentagons and a hexagon in fullerenes.
There have been many theoretical discussions as to whether there would be any electron delocalisation (i.e. aromaticity) at all in fullerenes, e.g. [2]; an unambiguous conclusion was obtained through hydrogenation (Section 3.1).
3 Reduction–formation of fullerene hydrides
The reduction of [60]fullerene by sodium and liquid ammonia gave the addition of 36 and 18 hydrogens [3]. The proposed Th symmetry structure for C60H36 was most improbable, requiring total non-conjugation in the product, and was disproved later (Sections 5.2 and 5.4).
Hydrogenation of [60]fullerene in benzene using hydrogen at atmospheric pressure with catalysts such as Pt/C and Pd/C, gave a solid on removal of the solvent from the resultant yellow solution that would only redissolve in aqueous acetone. Reduction by hydrogen transfer from cyclohexadiene gave the same result. (Hydrogen transfer was concurrently being investigated by other workers [4].) The cause was the rapid allylic oxidation of hydrofullerenes to fullerenols (see also [5]). Cage reduction could not be effected with Raney nickel catalyst [6], a feature used in some cycloadditions described below (Section 9).
3.1 Reduction by di-imide
Using reduction by di-imide produced all eight possible isomers of C60H4, and C60H2 [6] (also produced by reduction via hydroboration [5] and hydrozirconation [7]). (The assignment of singlets due to the 1,2,18,36- and 1,2,55,60 isomers, was updated later [8].)
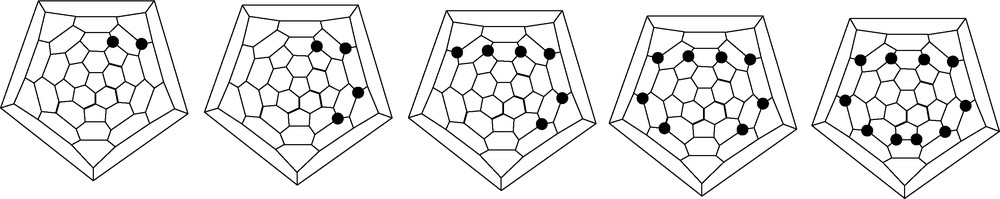
Some 30% of the tetrahydro[60]fullerene mixture consisted of the 1,2,3,4-isomer (Fig. 2). If there was no delocalisation in [60]fullerene, the π-density of a (localised) double bond in any hexagon would not be altered significantly by this addition. Hence subsequent addition should occur equally at any of the remaining eight (different) bonds (except for the double bond opposite on the cage which is statistically disadvantaged by a factor of 4:1). The preferential formation of the 1,2,3,4-tetrahydro derivative showed the cage to be partly aromatic. Addition in one hexagon causes localisation of the remaining electrons in that ring so favouring further addition there. This pattern is found in a number of other reactions. Extension of the arguments predicted that further addition should give rise to the ‘S’ and ‘T’ patterns (Fig. 3) [9] observed for example in epoxide formation [10].

Schlegel diagram of the principle (1,2,3,4) isomer of tetrahydro[60]fullerene.
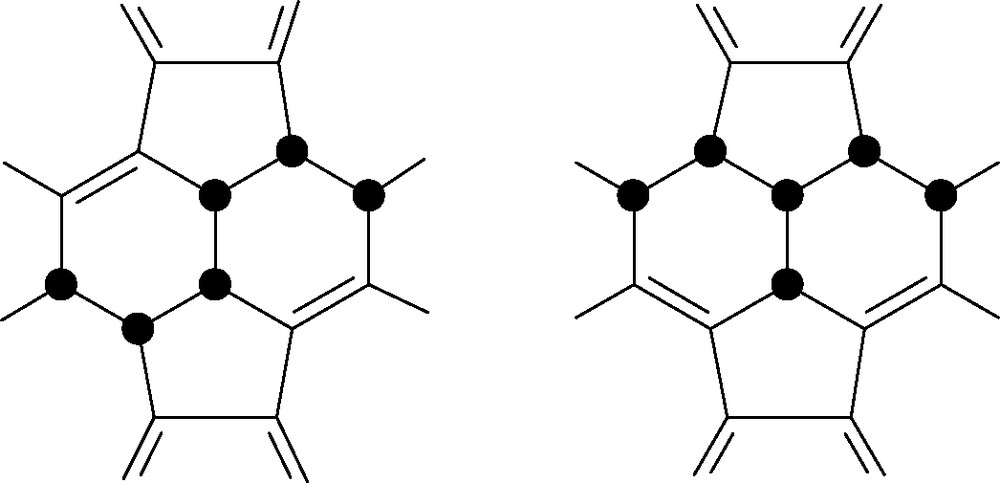
Localisation-driven ‘S’ and ‘T’ addition patterns for fullerenes; ● = addend.
Di-imide reduction of [70]fullerene produced 1,2- and 5,6-dihydro[70]fullerenes (Fig. 4) in a 25:1 ratio [6], cf. 2:1 for hydrogenation via hydroboration [11], demonstrating the greater reactivity of the 1,2- compared to the 5,6-bond [12]. The 5,6-bond has the slightly higher π-bond order [13] but greater curvature favours addition across the 1,2-bond. The 1H-NMR signal for the 2-hydrogen (attached to the more electronegative a-carbon [14]) is 0.5 ppm downfield relative to that for the 1-hydrogen (attached to a b-carbon), a feature used in characterising fullerene derivatives.

Structures of 1,2- (left) and 5,6-dihydro[70]fullerenes (right).
Further di-imide reduction of [70]fullerene gave six tetrahydro[70]fullerenes, the 1,2,3,4 and 1,2,5,6 isomers (Fig. 5) each comprising about 25% of the total due to the effect of π-bond localisation. The more equal amounts of the two main isomers compared to the dihydro precursors is a statistical effect.
1,2,3,4- (left) and 1,2,5,6-tetrahydro-[70]fullerenes (right).
3.2 Reduction by Zn/HCl
[60]Fullerene in e.g. benzene is rapidly reduced by Zn/HCl, a method that gives no byproducts; C60H36 is produced which on strong heating degrades C60H18. As in all other reactions, [70]fullerene is less reactive than [60]fullerene and gives C70H36, C70H38 (main) and C70H40 [15], as it does in fluorination (Section 5.2).
Reduction of the higher fullerenes yielded C78H36–48 and C84H48–52 as major components [16]. At higher DCI probe temperatures (EI ms) the C84 derivative gave a strong signal for the (less volatile) C84H40 showing it to be particularly stable (cf. fluorination, Section 5.3).
Degradation of the higher fullerenes occurs to give hydrides of the lower one and methylene derivatives, presumably from cage degradation. Formation of methylene derivatives is a common feature of many fullerene reactions, e.g. [17].
Reduction with Zn/DCl gave higher addition levels, i.e. up to C60D44 and C70D48, attributed to the greater stabilities of C-D vs C-H bonds providing greater compensation for the increased steric compression and loss of (aromatic) resonance energy that accompanies further addition.
Calculations [18] supported our proposal that C60H36 contains four planar aromatic rings (T symmetry, Fig. 6) due to the removal of strain from the adjacent pentagons [19]. Indirect proof was obtained from fluorination (isostructural with hydrogenation, but not subject to oxidative instability) which showed that C1 and C3 isomers were also present, Section 5.2). Recently these isomers were isolated in hydrogenation (the assumed structure of the C1 isomer being slightly in error by virtue of a 1,3-hydrogen shift) [20].
Schlegel diagram showing T symmetry C60X36 (● = H or F).
The best hydrogenation method is to heat fullerenes with hydrogen under pressure, control of the hydrogenation level depending upon temperature and pressure. C60H18 made in this way has the crown structure (Fig. 7, ● = H) [16] predicted by calculations [21] with a central planar aromatic ring. The structure is a sub-set of T-symmetry C60H36 and contains a threefold combination of either of the ‘S’ or ‘T’ patterns (Fig. 3).
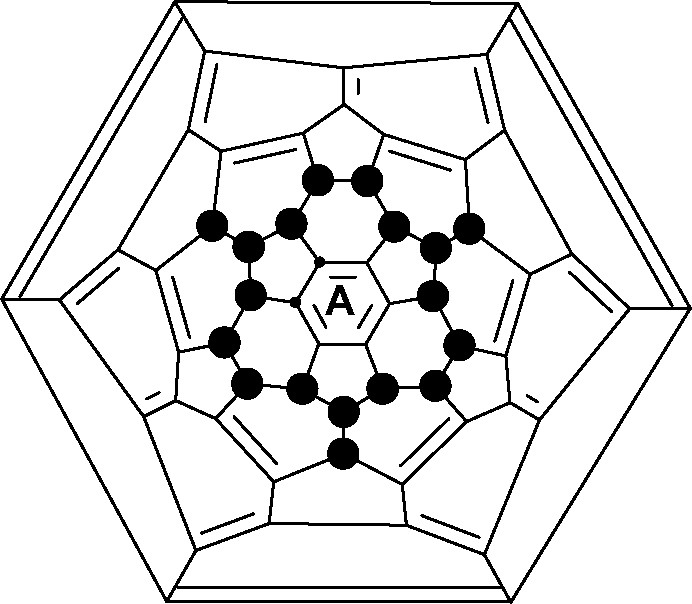
Schlegel diagram of C60X18 (● = H, F), showing the central aromatic ring.
Reduction of (C59N2) with Zn/HCl produces C59NH5, the conjectured structure being shown in Fig. 8 [22]. This is stable due to the formation of a 6π-electron (aromatic) pentagonal ring, consequently there is a high energy barrier to further reduction. This result contrasts with that observed in fluorination (Section 5.5).
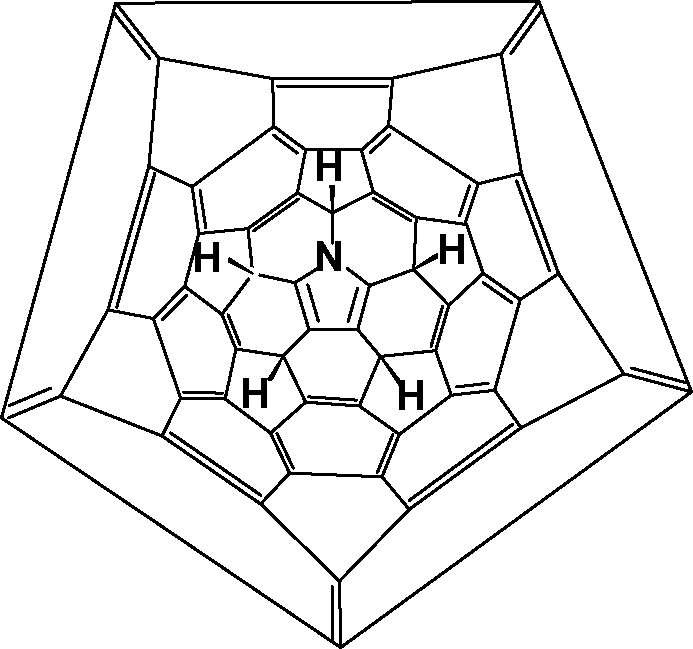
Schlegel diagram of conjectured structure for C59NH5.
3.3 Reduction by hydrogen sulphide
If traces of CS2 used for extracting fullerene soot are not completely removed, then on passing down an alumina column (containing traces of water), this becomes converted into H2S, COS, CO and CO2 (a little-known reaction [23]). The H2S then reduces the fullerene and is converted into sulphur (Scheme 1). Separate experiments showed that C60 converts H2S into sulphur [24].
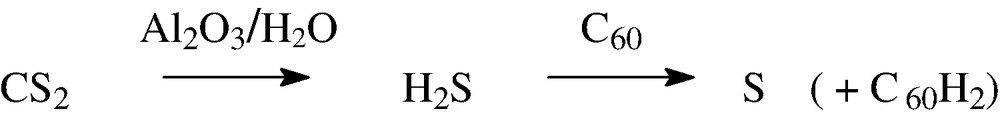
[60]Fullerene-catalysed conversion of CS2 into sulphur.
4 Hydroboration
Hydroxyfullerenes (fullerenols) are water soluble, and can be converted into a variety of polymers with very strong cross linking. They are made by nitration followed by substitution of NO2 by OH [25], and also by hydroboration (reaction with BH3/THF complex followed by H2O2/NaOH) [26]. The ready occurrence of allylic oxidation means that multiple addition of H and OH pairs in the latter reaction (Scheme 2) can only be achieved if the reaction is performed under N2. Otherwise the product shows an IR spectrum similar to that obtained by the nitration route. Allylic oxidation is also responsible for a typical fullerenol IR spectrum being obtained on quenching the organoborane intermediate with glacial acetic acid.
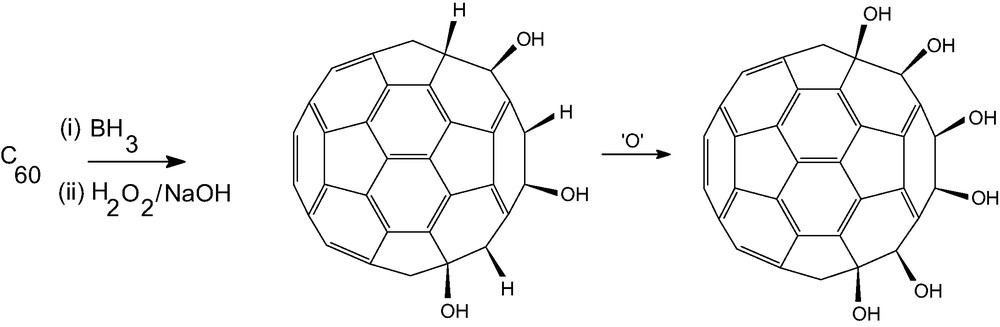
Representation of fullerenol formation via allylic oxidation of hydroborated intermediates.
5 Fluorination
Both fluorination and hydrogenation have low steric requirements, consequently give similar polyaddition patterns. However, the low activation energy for fluorination makes the reaction very hard to control. Fluorination has been reviewed recently [27].
5.1 Fluorination by F2,F2-inert gas complexes, and by interhalogens
Initial experiments used pure samples of [60]fullerene fluorinated by either F2 gas or XeF2 at 70 °C. The slow fluorination rate (many days) [28] contrasted with another report [29] because pure [60]fullerene (then unavailable elsewhere) in which the molecular close packing restricts lattice penetration of fluorine, was used. This also explains why reaction of [60]fullerene with fluorine gas at 70 °C for 24 h, gives a product consisting of a mixture of highly fluorinated- and unreacted fullerene [30] The packing in [70]fullerene is also poorer than in [60]fullerene, consequently it fluorinates faster; [30] in other reactions it is less reactive. The mass spectra of the products show a very wide distribution of fluorinated species making isolation of individual components by this route unfeasible.
The hopes that fluorofullerenes would be superlubricants [31], neglected their inability to assume the eclipsing-avoiding twisted backbone of Teflon, so are very susceptible to nucleophilic substitution. Dry solid fluorofullerenes are stable and hydrophobic, but addition of a cosolvent to water e.g. acetone, gives an immediate exothermic reaction evolving HF and formation of epoxides [32]. Fluorofullerenes are therefore lubricants but can, however, be useful synthons, e.g. reaction with various carbon nucleophiles [32]. Given that an SN2 reaction is sterically impossible a SN2′ mechanism was proposed (Scheme 3, see also Section 7).
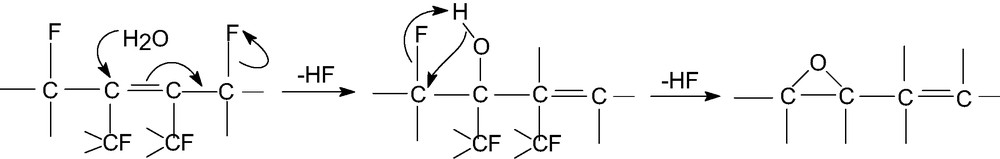
Mechanism for nucleophilic substitution of fluorofullerenes, followed by epoxide formation.
In the spectrum of the product of fluorination of [60]fullerene, species corresponding to C60F18 and C60F36 were dominant, providing the first indication that the regiochemistry of fluorination parallels that for hydrogenation [32]. CF3 groups are formed through fragmentation, and CF2-containing species are present, attributed to cage fragmentation followed by addition to another cage (cf. hydrogenation, Section 3.2).
The lability of bromine and chlorine in fullerenes [33,34] was employed to limit the extent of fluorine addition i.e. fluorinate a halogenofullerenes and then dehalogenate by heating. Though the derivatives tended to form complex degradation products, isolation of the mixed halogenated fullerene C60F14Cl18 indicated that the differing halogen sizes would permit formation of compounds of general approximate formula C60F48–2nCln [35].
C60F48 obtained by high temperature, was the first fluorofullerene to be characterised [36–38]. It forms a red crystalline charge-transfer complex with toluene, and deeper coloured complexes (bathochromic shift) with more alkylated benzenes [37]. C60F48 can also be produced by fluorinating with KrF2 [39]; hyperfluorination, which requires cage-opening, also occurs giving up to C60F78. Hyperfluorination occurs during fluorinating with F2 under UV irradiation (giving C60F76) [40], and with TbF4 (giving C60F70) [41].
5.2 Fluorination with transition metal fluorides
5.2.1 [60]Fullerene
Fluorination with transition metal fluorides was introduced by DuPont in 1947, fine control being achieved by adjusting the fluorine-release temperature [42]. The method is ideal for use with involatile solids such as fullerenes, especially as the fluorofullerenes are more volatile, and under vacuum these can be swept away from the reaction zone so that limited fluorination can be achieved [43]. By selection of the metal fluoride and reaction temperature, specific derivatives can be obtained. The high polarity of the C-F bond and the solubility of fluorofullerenes, allows HPLC purification and separation of species of identical fluorine content.
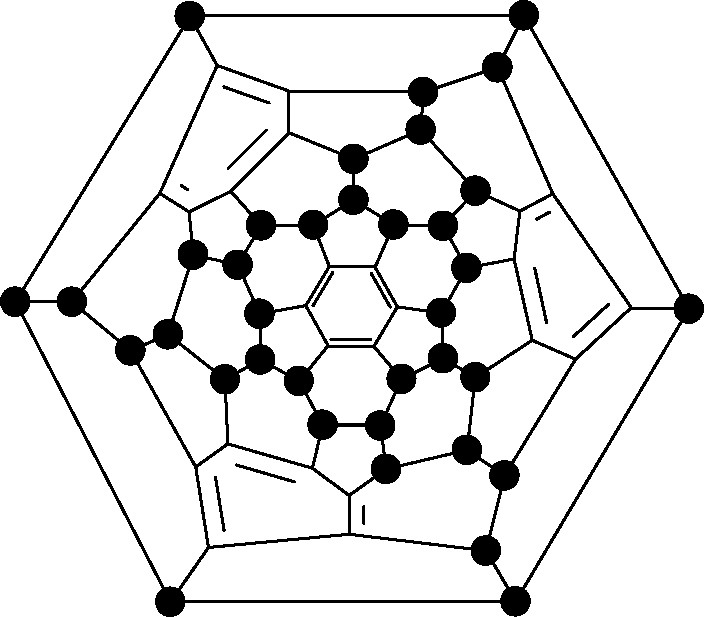
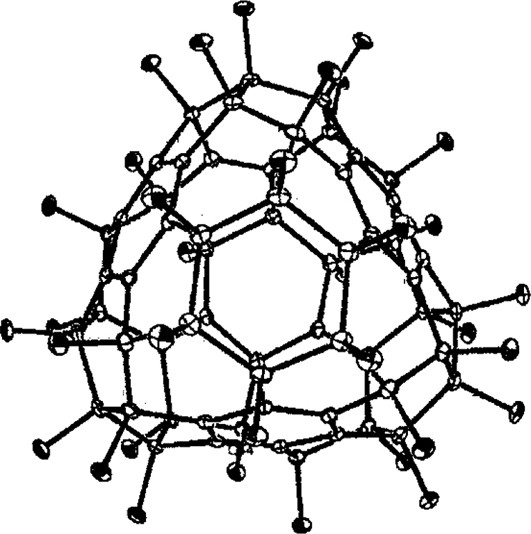
Reaction with K2PtF6 produced fully characterised C60F18 (Fig. 7, ● = F) [44]. The concentration of the fluorines at one end of the molecule (shown by the single-crystal X-ray structure, Fig. 9 [45]) gives it a very high dipole moment (ca. 6 Debye) [46] and a very long HPLC retention time. Reaction proceeds via cascade processes (see Section 3.1).

X-ray structure for C60F18.
C60F18O (Fig. 10) was the first oxahomofullerene (fullerene ether) to be isolated and characterised [47]; other fluorofullerene ethers and bis ethers have been obtained subsequently [48].
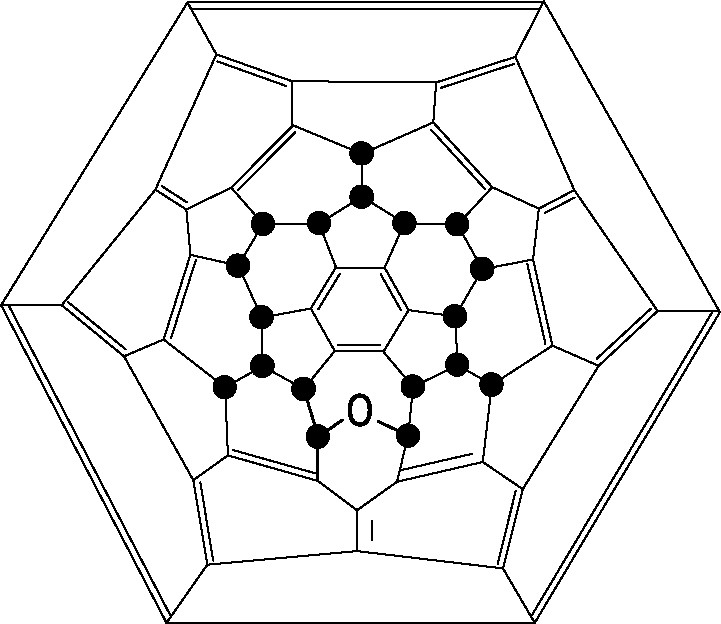
Schlegel diagram of C60F18O; ● = F.
As well as the T isomer of C60F36 (Fig. 6), the X-ray structure of which showed it to be the most distorted fullerene (Fig. 11) [49], the C3 and C1 isomers have also been isolated from fluorination with MnF3 (Fig. 12). All three differ only through 1,3-fluorine shifts [50,51]; the T isomer contains four planar aromatic rings whilst the other isomers each have three.
X-ray structure of T-C60F36.
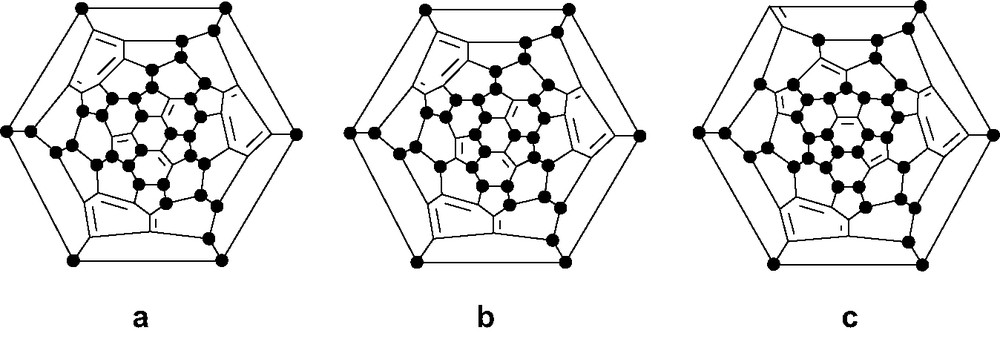
Schlegel diagrams of the C3 and C1 isomers of C60F36, and the C1 isomer of C60F38.
Fluorination with TbF4 is more aggressive and gives mainly C60F42 and C60F48 at 320 °C and 350 °C, respectively [52]. Partially characterised compounds C60F26, C60F28, C60F30 and C60F22 have been isolated from fluorination with MnF3 [53], whilst the same reagent gave fully characterised C60F38. Very complex analysis of the spectroscopic details revealed the latter structure to be that shown in Fig. 12c [54], whereby disruption of one of the aromatic rings of C60F36 occurs on addition of two fluorine atoms.
The lowest fluorinated species C60F2 has also been isolated along with C60F4, C60F6 and C60F8, this series demonstrating the sequential 1,2-addition pattern, but also a departure to a 1,4-addition pattern in C60F8 (Fig. 13) [55,56]. Related patterns have been obtained in the series C60FnO, n = 2, 4, 6, 8, 10 [56,57].
Motifs for C60Fn (n = 2, 4, 6, 8).
Other novel product is C60F16 which differs from C60F18 only in the absence of two fluorines that lie along one of the symmetry planes [58]. But why the loss of only two fluorines, i.e., one could expect to find C60F14 and C60F12? Furthermore, the HPLC retention time is more than double that of C60F18, whilst that of C60F16O (ca. 150 min, [59]), grossly exceeds that of any other fluorofullerene. The existence of dimers cannot be discounted, especially as C60F16 is readily made by pyrolysis of C60F18 [60].
C60F20 (‘Saturnene’) is unique in having all equatorial fluorines creating a flattened structure (Fig. 14) [61].
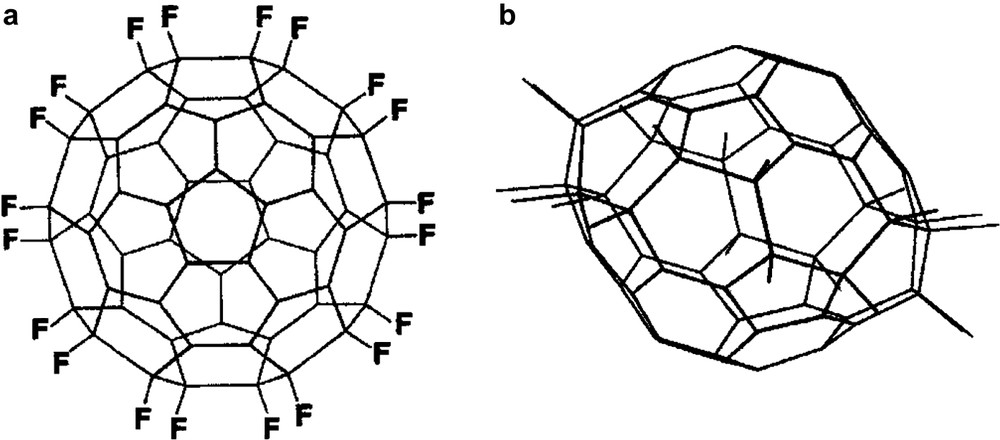
Structure of C60F20 (‘Saturnene’).
Fragmentation of the fluorofullerene cages accompanies fluorination, producing : CF2 carbenes which can insert into C–F bonds giving derivatives such as C60F17CF3 (Fig. 15) and C60F17CF2CF3 one isomer of the former being chiral [62]. Up to threefold addition should be possible but there is only scant spectroscopic evidence for this.
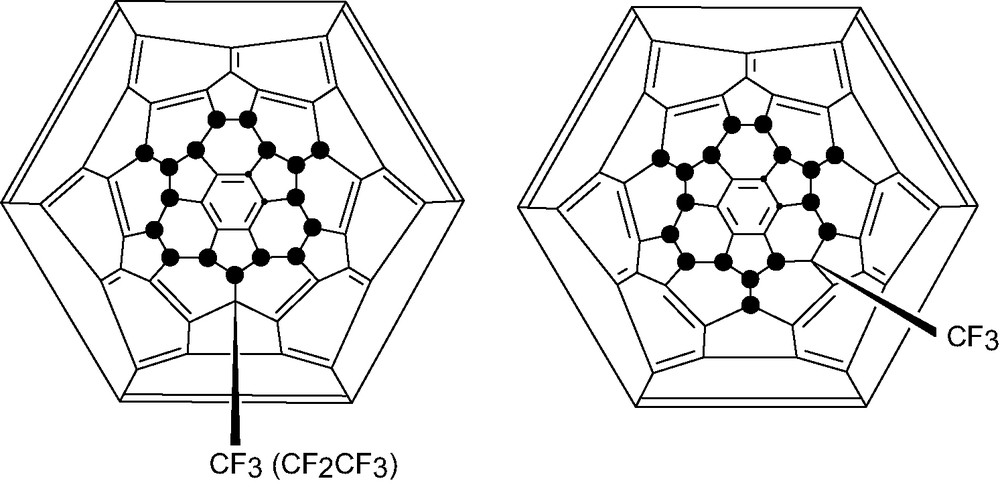
Schlegel diagrams for C60F17CF3/C60F17CF2CF3).
5.2.2 [70]Fullerene
The main product of fluorination with MnF3 is C70F38 together with smaller amounts of C70F36 and C70F40, so paralleling hydrogenation. These components (and numerous isomers) have been isolated by HPLC; there are marked differences in polarities and symmetries (variously Cs/C2 and C1) of some [63]. Our proposal that the specific addition levels observed indicated aromatic structures [64] has been confirmed by X-ray characterisation of the C2 and C1 isomers of C70F38 (Fig. 16, which shows the distortion) These contain 3- and 4-planar aromatic rings, respectively, cf. C60F36 [65].

C2 (left) and C1 (right) C70F38; the end-on view of the former illustrates the distortion.
5.2.3 Higher fullerenes
Fluorination of higher fullerenes by MnF3 and CeF4 yields C76F38, C78F38, C82F40, C84F40 and C84F44, with C84F40 being by the most abundant species [66], paralleling the high stability noted above (Section 3.2) for C84H40. The structures of C84F40 (Fig. 17) and C84F44 are cubic [67].

The cubic structure of C84F40.
5.4 Fluorination of i-3He[60]fullerene
The shift in location of the single line in the 3He-NMR spectra of fullerenes containing 3He within the cage is a measure only of the change in aromaticity of the cage surface. The 3He-NMR spectra of i-3HeC60F18 and i-3HeC60F36 are virtually identical to those of the corresponding hydrogenated species thereby confirming their isostructural relationships [68]. Moreover, the spectra for T and C3 isomers of C60X36 consist of two lines in ca. 1:3 intensity ratio, the chemical shifts relative to the parent fullerene being predicted accurately [68].
5.5 Fluorination of C59N
Whereas hydrogenation of C59N produces H5C59N (Fig. 8), fluorination by MnF3 produces C59NF33 [69]. As noted (Section 3.2) hydrogenation of C59N creates an increase in aromatic stability, so reaction does not readily proceed further. However, the much lower activation energy for fluorination overcomes the barrier to further reaction.
In e.g. T C60F36 (Fig. 6) many pentagons containing three fluorines. If two fluorines are removed from one of these rings and the remaining C–F bond is replaced by N, this creates a 10π aromatic circuit and enhanced aromatic stability (Fig. 18).

Conjectured structure for C59NF33 based on T C60F36 (● = F); similar structures can be obtained from the other C60F36 isomers.
Other species isolated from this work include C59NF, C59NF17, C59NF37, C59NF4CF3, C59NF3(CF3)2 and C59NF2(CF3)3 and structures for these have been proposed.
6 Bromination and chlorination
These radical reactions have steric requirements different from fluorination resulting in quite different regiochemistry, with 1,4-addition being a dominant feature.
6.1 Formation of C60Cl6 and C70Cl10
Chlorination of [60]- and [70]fullerenes by ICl gives C60Cl6 and C70Cl10, respectively (Fig. 19) [70,71].
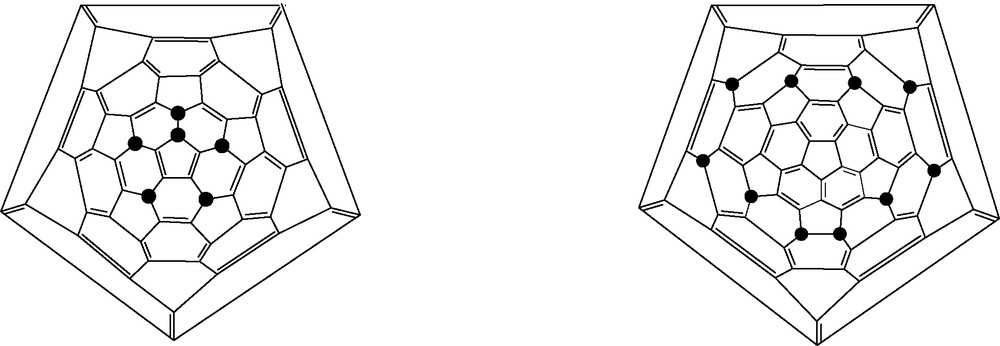
Schlegel diagrams for C60Cl6 (left) and C70Cl10 (right); ● = Cl.
Since the end cap motif of [70]fullerene is isostructural with that in [60]fullerene, chlorination could have been expected to give C70Cl6. The formation of C70Cl10 was attributed to the unfavourable bond disposition around the equator of [70]fullerene due to the need to have double bonds exocyclic to the pentagons [71]. It can be ameliorated by consecutive 1,4-additions around the equator, resulting in two adjacent chlorines as in C60Cl6; the resultant steric compression creates regions of high reactivity. The structure is calculated to be of low energy [72] and many other isostructural compounds are now known.
6.2 Formation of C60Br24, C60Br8 and C60Br6
Bromination of [60]fullerene by bromine gives either C60Br24, C60Br8 or C60Br6 (isostructural with C60Cl6) depending upon the solvent employed [73,74]. The motif in C60Br6 is found in many other derivatives, whereas the C60Br8 motif has been found only in C60Me8 [75]. C70Br10 has been isolated from bromination of [70]fullerene (cf. Section 6.1) [76] (Fig. 20).
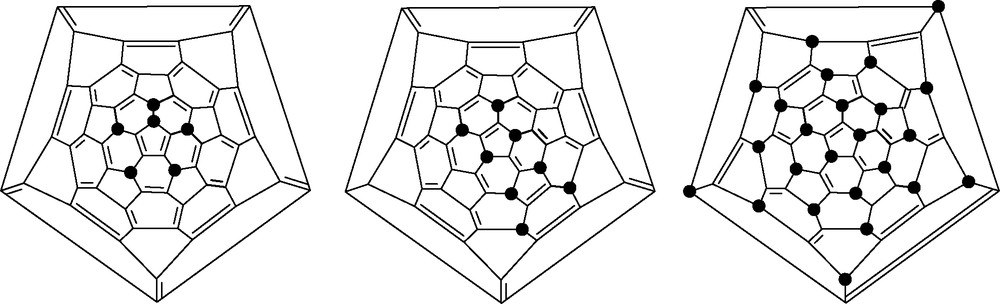
Schlegel diagrams of the structures of C60Br6 C60Br8 and C60Br24 (● = Br).
7 Nucleophilic substitution–electrophilic aromatic substitution
Nucleophilic substitution follows two mechanisms: in the absence of a Lewis acid catalyst, an SN2′-type of mechanism must apply. In the presence of a catalyst, the mechanism proceeds via the formation of an intermediate cation (or partial cation) feasible here due to the strong electron-withdrawing property of the cage being reduced by the presence of less electronegative sp3-hybridised carbons [77]. When the nucleophile is an aromatic compound, the reaction becomes an electrophilic aromatic substitution by the halogenofullerene.
7.1 Nucleophilic substitution of fluorofullerenes
7.1.1 Reaction of fluorofullerenes with water
A range of specific derivatives of fluorofullerenes have been isolated. Replacement of F by OH is followed by elimination of HF, and this can be repeated. Further elimination involving loss of CO from the epoxides may occur, giving species such as C59F34 [78]. In a more detailed investigation specific C1 isomers of C60F35OH have been obtained from C3 and C1 C60F36 [79]. This work identified the motif required for ready substitution, viz. the presence of two CF bonds adjacent to at least on end of a double bond (Scheme 3), so that T C60F36 is unreactive and likewise is C60F18. The motifs in C60F48 require that only 12 fluorines can be replaced, so giving it a relatively high hydrolytic stability [80].
7.1.2 Reaction of C60F18 with bulky carbon nucleophiles – formation of trannulenes by the SN2″ reaction
Reaction of bulky carbon nucleophiles with C60F18 leads to the formation of trannulenes, a new class of compounds having around the equatorial region, a fully delocalised 18π aromatic chain with all bonds of equal length; the compounds are bright emerald-green, are exceptionally stable and have a barrell-like structure (Fig. 21) [81]. This unique organic reaction, defined as an SN2″, (or extended SN2′) reaction, involves an incoming nucleophile displacing a fluorine at the δ-position. Two further types of trannulenes have since been discovered in other reactions [82].
Schlegel diagram (left; dotted line is delocalised chain) and X-ray structure (right) of a trannulene.
7.1.3 Reaction of C60F18 with aromatics—the ‘triumphenes’
In the presence of a FeCl3, either one, two, or three of the outermost fluorines of C60F18 can be substituted by aromatics (Fig. 22), an electrophilic substitution of the fluorofullerene into the aromatic. Hence more reactive aromatics are substituted more readily [83]. A very surprising result was the formation of C60Ph18, the most phenylated organic compound known. The phenyl groups are almost certainly located in the positions formerly occupied by fluorine, creating an exceptional structure.
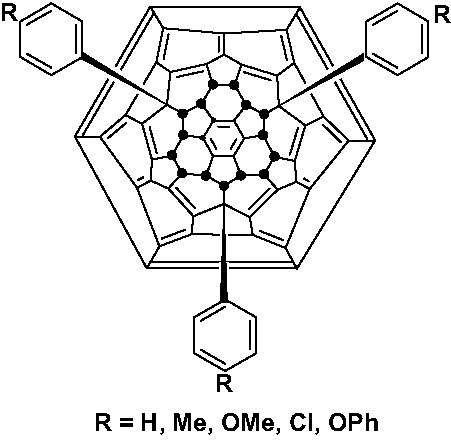
Schlegel diagram of tris-substituted C60F15Ar3 derivatives.
Electrophilic substitution of C60F18 into phenols occurs at both ortho and para positions. The former products then undergo 1,3-fluorine migration being followed by HF elimination between the phenolic OH group giving benzo[2′,3′:10,11]hexadecafluoro[60]fullerene (Fig. 23) and derivatives; complete defluorination can also occur [84].

Benzofurano[2′,3′:10,11]hexadecafluoro[60]fullerene from C60F18 and phenol.
7.2 Chlorofullerenes as electrophiles
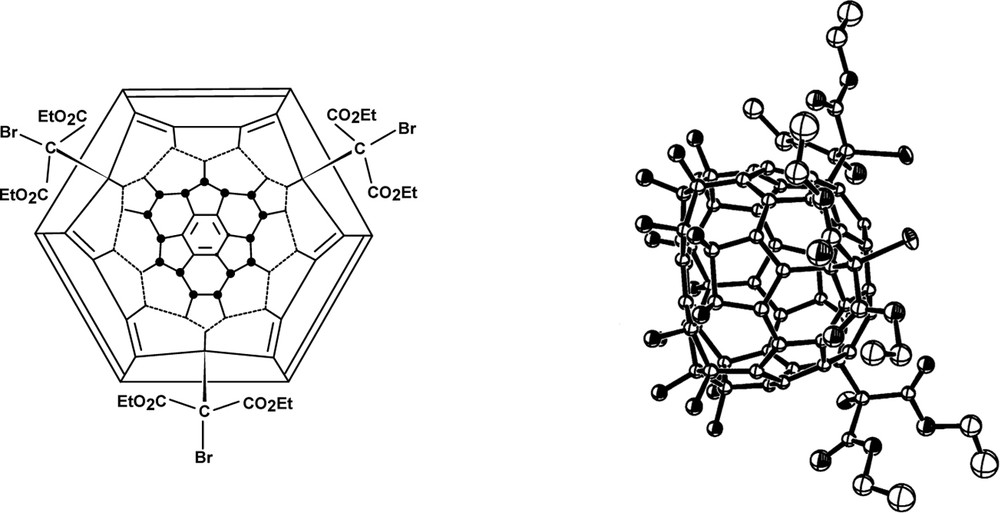
Reaction of C60Cl6 with benzene/FeCl3 produces C60Ph5Cl (Fig. 24, X = Cl), which on reaction with PPh3/H2O yields C60Ph5H (Fig. 24, X = H) [85]. Other aromatics give the corresponding Cs symmetric derivatives [77], and as in the case of aryldefluorination above, steric hindrance causes either largely or exclusively para substitution in the aromatics. Steric hindrance prevents mesitylene from reacting, and likewise causes substitution at the para position in trimethylsilylbenzene instead of cleavage of the trimethylsilyl group (ipso substitution), normally the much more facile reaction [86].
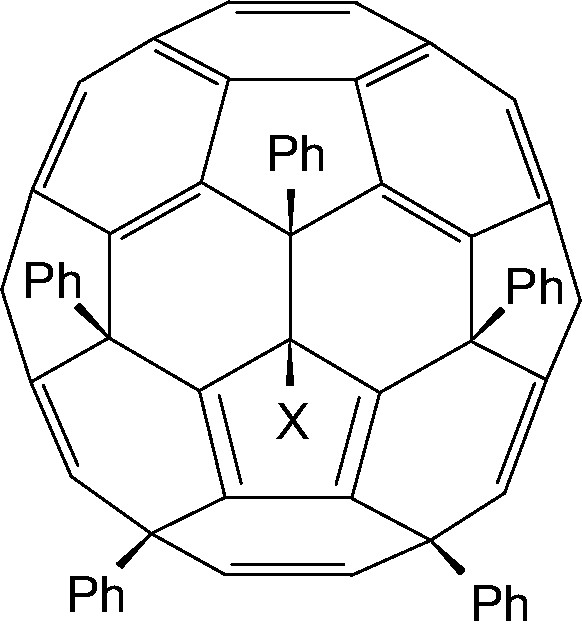
Structure of C60Ph5X (X = Cl, H).
The entering aryl groups occupy the positions of the departing chlorines showing reaction to occur via an intermediate cation proved by the isolation of C60Ph5+ [77]. Removal of the chlorine creates an anti-aromatic structure, so a 1,2-shift of an aryl group occurs and probably is synchronous with the departure of the chlorine (Scheme 4). The location of the positive charge is confirmed by single crystal X-ray structures of derivatives formed by reaction with nucleophiles e.g. CN– (from Me3SiCN) [87]; a further rearrangement must also occur since a second unsymmetrical uncharacterised derivative is formed. Chemical shifts (13C-NMR) for the on-axis carbons on the cage opposite to the cation, indicate endohedral homoconjugation.
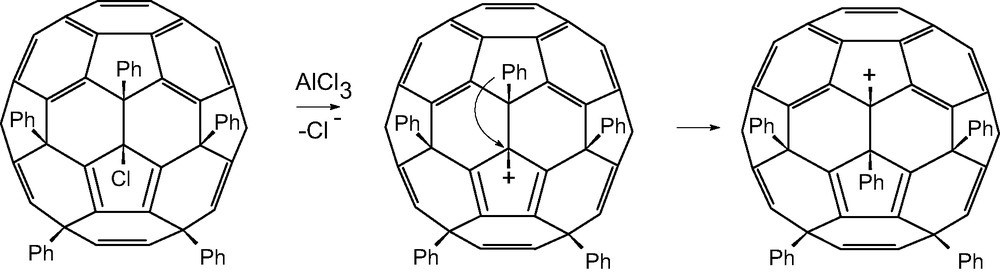
Formation and stabilisation of a penta-aryl[60]fullerene cation.
Replacement of the sixth chlorine by aryl is sterically hindered, so that although attack by aryl at the carbocation in the right hand structure of Scheme 5 gives Cs C60Ph6, an unsymmetrical isomer is also obtained; 2-D and temperature-dependent NMR studies indicate the presence of three adjacent phenyl groups in this [88]. By contrast, reaction with the less sterically demanding allyl group (from allyltrimethylsilane catalysed by TiCl4) readily gives Cs C60(allyl)6 and C60(allyl)5Cl [89].
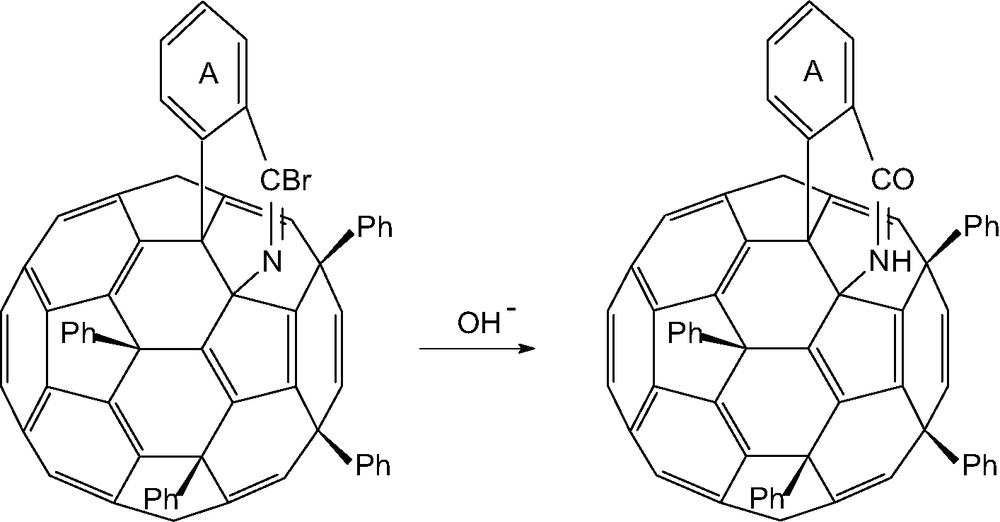
Formation of an isoquinolono[60]fullerene from reaction of C60Ph5Cl with CNBr/FeCl3.
Symmetrical C60Ph2, unsymmetrical C60Ph4 (Fig. 25), and traces of C60Ph3H3 are obtained as by-products from the C60Cl6/benzene/FeCl3 reaction; [90] the formation of hydro derivatives is a common feature of many fullerene reactions (see e.g., Section 7.3). Pathways involving either 1,2-, 1,4-, or 1,6-Cl2 loss from either the precursor or partially phenylated intermediates can be envisaged for the formation of the main products. This combination of elimination and nucleophilic substitution of chlorine is also found in reactions with alkoxy nucleophiles [91] and in the reactions of C70Cl10.
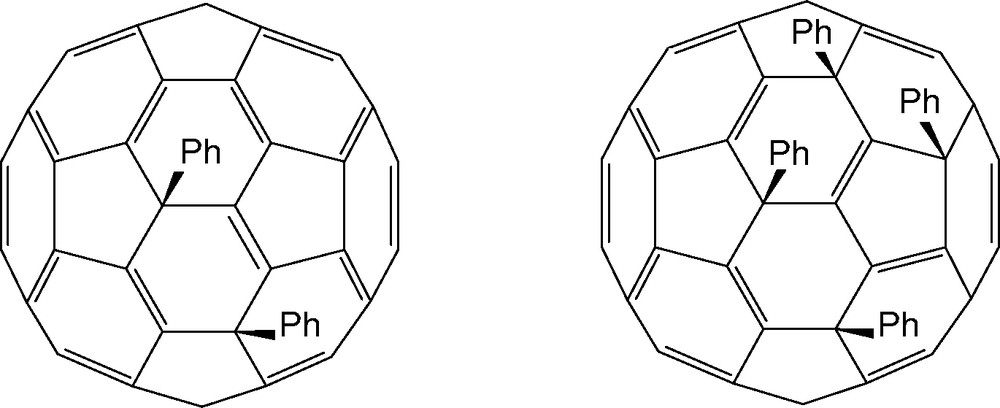
Structures of symmetrical C60Ph2 and unsymmetrical C60Ph4.
The reaction between C60Ph5Cl and CNBr/FeCl3 produces a bromoisoquinolino-[60]fullerene due to nucleophilic replacement of the chlorine by N≡C–Br, followed by electrophilic substitution into the ortho position of the adjacent phenyl ring (Scheme 5); this latter is inhibited by electron withdrawal in the aryl ring, hence the corresponding fluorophenyl derivatives is unreactive. Subsequent reaction with hydroxide ion occurs readily and gives isoquinolono[3′,4′:1,2][60]fullerene [92]. Reversal of the positions of attachment of the nitrogen and the aryl group A cannot be excluded.
A related ring closure involves conversions of C70Ph5H into C60Ph4C6H4O2 a benzo[b]furano[60]fullerene (Fig. 26) [93,94]. Facile spontaneous oxidation of the C–H bond to C–OH creates steric compression with the adjacent aryl group, so oxidative coupling occurs. The oxides undergo fragmentation under EI mass spectrometry to give C58 derivatives. Formation of benzofurano[60]fullerenes likewise occurs spontaneously from aryloxy[60]fullerene precursors (obtained from reaction of phenols with C60Cl6 [95], a reaction that parallels the reaction of phenols with C60F18 (Fig. 23).
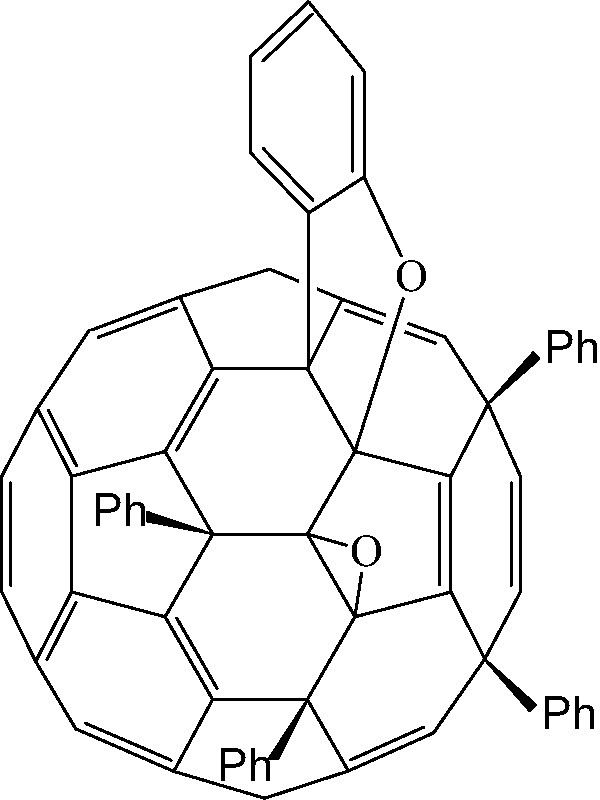
A di-epoxy derivative of a benzo[b]furano[60]fullerene.
Reaction of C70Cl10 with benzene/FeCl3 produces C70Ph8 and C70Ph10 as major products (Fig. 27) and C70Ph2/4/6 as minor ones [96]. C70Ph10 is not formed directly from the chloro precursor, but is produced by phenylation of C70Ph8 (which is formed more readily) so the adjacent and eclipsed chlorines are preferentially lost during the reaction, as for example they are in alkoxylation of C60Cl6; this reaction sequence means that the final two aryl groups can be made to differ from the others, simply by reacting C70Ph8 with ArH/FeCl3.
Schlegel diagrams for C70Ph8, C70Ph10 (● = Ph), C70Ph9OH (X = Ph), and C70Ph8(OH)2 (X = OH).
Minor products from the above reaction are C70Ph9OH, the first monohydroxyfullerene to be isolated, and C70Ph8(OH)2 (Fig. 27) [97]. The mechanism of their formation is unclear.
A unique reaction occurred on allowing C70Ph8 to stand in light, whence it changed into a bis lactone, C70Ph8O4 which has an 11-member hole in it (Fig. 28), the first [70]fullerene derivative to have an opened cage [98].
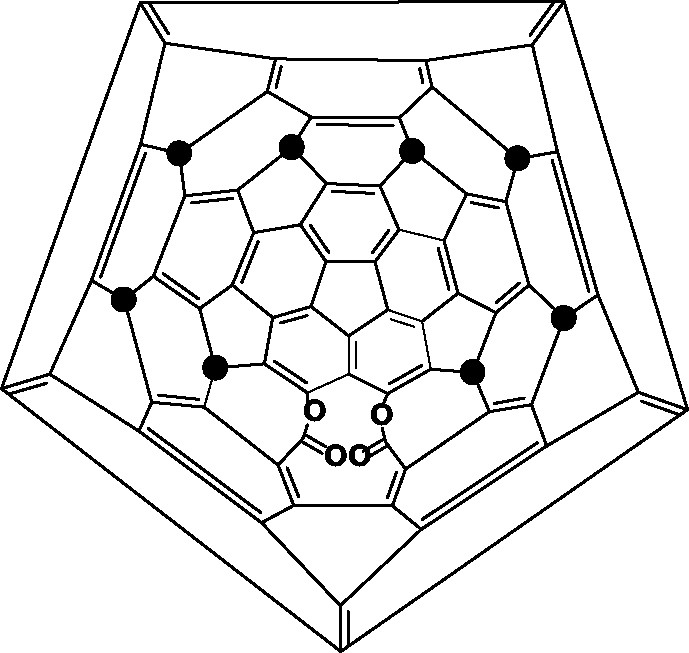
Structure of the spontaneously formed bis lactone, C70Ph8O4; ● = Ph.
7.3 Bromo[60]fullerenes as electrophiles
Heating a solution of benzene, bromine (which increases the electrophilicity of the cage), and FeCl3, gives many products including: C60Ph5H (main product), C60Phn (n = 4, 6, 8, 10, 12), C60PhnO2 (n = 4, 6, 8, 10, 12), C60PhnOH (n = 7, 9, 11), C60Phn = 4, 10), C60Ph4H4, C60Ph5H3, C60PhnO2H (n = 5, 9), C60Ph4C6H4O2 [formed by spontaneous oxidation of C60Ph5H (see Section 7.2)], C60Ph9OH3, and C60Ph11O3H2 [99,100]. The corresponding reaction with toluene and chlorobenzene produced as main products, C60(MeC6H4)4 and C60(ClC6H4)5H, respectively. However, specific derivatives are more easily obtained by reaction of chlorofullerenes (Section 7.2).
8 Formation and reactivity of oxides
Despite claims to the contrary, fullerenes do not occur naturally because of their high susceptibility to oxidation [101], the first fullerene reaction to be qualitatively observed. Films of pure fullerenes on rotovap flasks acquired a pinkish hue after a few days and would not then redissolve due to oxide formation [102]. This air oxidation is photocatalysed [103]; the product has been shown to be C120O (Fig. 29a), is present in all samples of C60, and is believed to be formed from a [2 + 2] reaction between C60 and C60O [104].
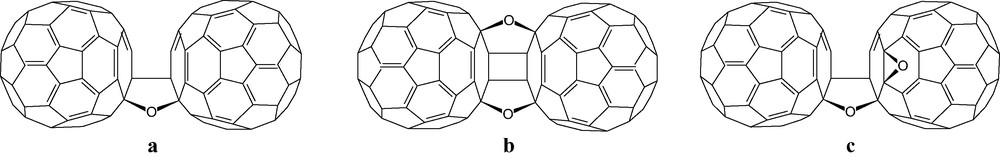
a, b, c Structures of C120O and major (b) and minor (c) isomers of C120O2.
The structures of C120O [105] and two isolated isomers of C120O2 (Fig. 29) [106] were determined by NMR. The structure of the main isomer (Fig. 29b) was deduced also by application of the principle of minimisation of double bonds in pentagons, and other geometrical constraints [107]. Semi-empirical calculations for C120O, C120O2, (and also C130O, C130O2, C140O and C140O2), confirmed that the assigned structures are the most stable ones amongst a number of possibilities [108]. The structures of C120O2 also follow the principle (Fig. 2) that a second addition will preferentially occur adjacent to the first.
Some higher fullerenes (especially [78]fullerene can undergo rapid spontaneous oxidation, to give completely insoluble material. The extreme insolubility was at first attributed to graphitisation [109] matrix-isolated CO2 was found to be produced (IR) on heating KBr discs. (This KBr/fullerene matrix is quite remarkable, the CO2 being stable indefinitely at room temperature, or at 225 °C.) Since C60O, C70O, C120O etc. behave similarly, the insoluble material consists of oxides (probably polymerised) [110]. The question as to how CO2 could be produced from a mono-oxide was answered by the isolation of C119, formation of which was consistent with Eq. (1) [110].
| (1) |
2C119 is more usually obtained by heating a mixture of C60 and C60O, which is believed to lose CO to give an intermediate carbene (Scheme 6), having a strained four-member ring, (or a diradical). This may then either add to a 6:6 bond [104] or insert into a 6:5 bond of [60]fullerene, such addition/insertions being common in fullerene chemistry [105]. These intermediates are then conjectured to undergo intramolecular [2 + 2] cycloaddition, the product that results from the 6:5 insertion being the major one [106] because it is less strained.

Loss of CO from C60O to give an intermediate carbene (C59) which then reacts with C60 to give C119.
Substituted fullerenes can lose both CO and CO2. For example heating KBr discs of C70F38O reveals the formation of matrix-isolated CO [110] and ms. shows the formation of C69F38; this species, and others produced from similar oxides, are particularly stable since the doubly charged ions have greatly enhanced intensities [111]; the reason for this stability is not yet understood.
Loss of CO or CO2 on heating can result in the formation of lower fullerenes, which must contain seven-member rings [99]. Thus loss of 2 CO2 from C70Ph8O4 and of 2 CO from C60Ph4O2 yields C68Ph8 and C58Ph4, respectively (Fig. 30). These products have adjacent five-member rings, but are allowable because the neighbouring seven-member rings and the presence of the sp3-hybridised carbons in the five-member rings combine to reduce strain.
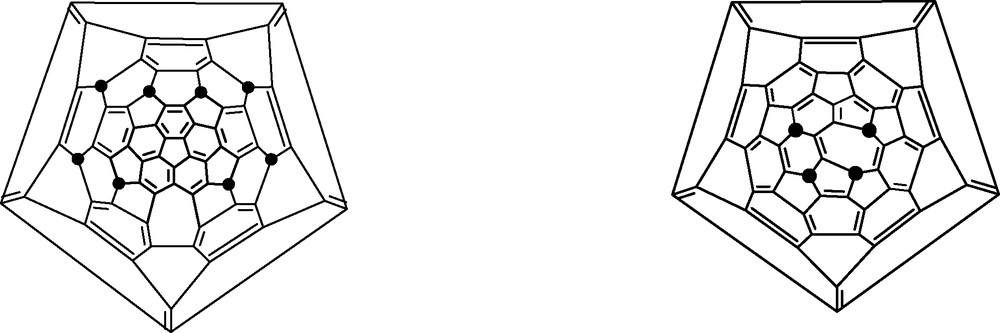
Conjectured structures of C68Ph8 and C58Ph4 (● = Ph).
[70]Fullerene epoxidises across the 1,2- and 5,6-bonds in a 43:57 ratio [112,113]. An unresolved general problem is why different reactions give such a wide variation in the relative proportions of the addition ratios [12] (see also Section 3.1). The epoxidation ratio is independent of formation during the arc-discharge fullerene production process [112] or by peracid oxidation of [70]fullerene [113] (Fig. 31).
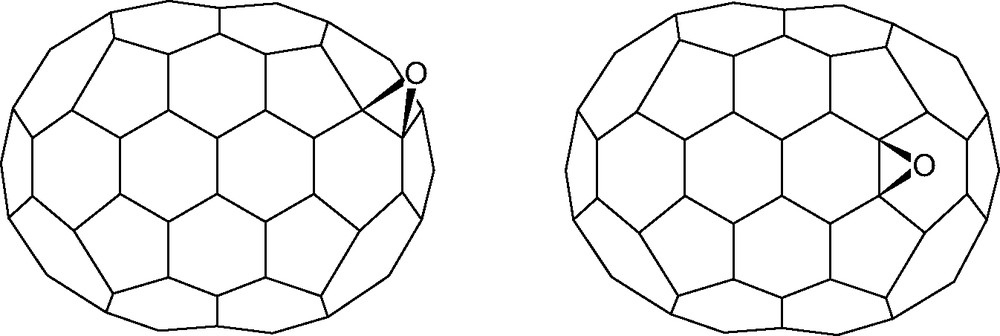
1,2-Epoxy[70]fullerene and 5,6-epoxy[70]fullerene.
9 Methylation
Methylation is important because although the introduced functional group is not very amenable to further reactions, the methylfullerenes are very much more soluble in a range of solvents, especially acetone and THF than the parent molecules [114–118]. The reaction can therefore be used to increase the solubility of other derivatives.
Methylation of [60]fullerene with Li/MeI gives C60Men (n = 2, 4, 6, 8, 10, 12, 14 and > 16) together with some oxide derivatives [114]. Both 1,2- Me2- and 1,4-Me2-C60 have been isolated and characterised (the former gives five oxide derivatives), along with C60Me8 which is isostructural with C60Br8 (Fig. 20), these being the only two derivatives having this motif. On reduction, 1,2-Me2C60 gives C60Me2H16, whilst 1,4-Me2C60 gives C60Me2H34 these results showing how important are the 18 And 36 site occupancies in [60]fullerene addition.
Reaction of C60Cl6 with MeLi yielded a range of fully characterised compounds (Fig. 32), and these confirmed the conjectured location of epoxy and OH groups obtained in related phenylated compounds [115]. Especially notable was the isolation of the bis-epoxide ketone (Fig. 33) a cage-opened tautomer of the bis-epoxide fullerenol, C60Me5O2OH [116].

Products obtained from reaction of C60Cl6 with MeLi.
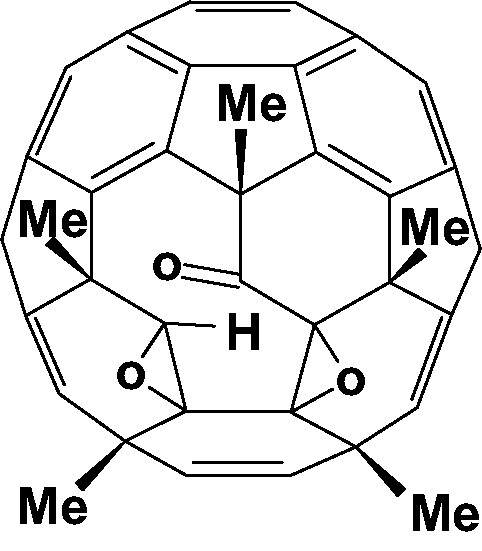
The cage-opened bis-epoxide ketone C60Me5O3H.
Methylation of [70]fullerene by Li/MeI gives mainly a 3.4:1 ratio of the 1,2- and 5,6-C70Me2 isomers (cf. epoxidation, Section 8) [114,117], accompanied by five others, one of which is probably the 7,21-isomer (obtained also by reaction of C70Cl10 with MeLi). Other uncharacterised products were C70Men (n = 4, 6, 8, 10), which may involve addition in the equatorial region. This was definitely shown to be the case on reaction of [70]fullerene with Al–Ni–NaOH in DMSO-THF followed by quenching with MeI. A sequential addition pathway from C70Me2 to C70Me10 occurs, starting with addition across the 7,23 bond, shown in Fig. 34, thereby confirming the previously conjectured pathway for phenylation. Just as C70Ph8 becomes oxidised on standing to the cage-opened bis lactone C70Ph8O4 so C70Me8 is converted to C70Me8O4, which is probably isostructural [117].
Stepwise addition pathway for methylation of [70]fullerene (● = Me, double bonds omitted for clarity).
Methylation of [76]fullerene by the Al-Ni-NaOH method gives mainly five isomers of C76Me2, one of which has been characterised as 1,6-Me2C76 together with some methyleneated compounds, C76(CH2)n (n = 2–4). The first oxahomo derivative of a higher fullerene 1,6-Me2C76O was also obtained (oxygen inserts into the 1,6-bond) and also the first bis-oxahomo derivative C2 C76Me2O2. [118]. Methylation of [84]fullerene takes place less readily, giving four dimethyl derivatives, one of C1 symmetry, the other being Cs or C2 [118].
10 Some cycloadditions
10.1 Benzyne
The [2 + 2] reaction of benzyne with [70]fullerene gave four monoadducts, whereas three had been observed in any other [70]fullerene cycloaddition [119]. These were characterised (1H-NMR) as 1,2-, 7,21-, 5,6-, and 7,23-benzeno[70]fullerenes [120], with a 1,2-/5,6-ratio of 3.5. Re-investigation involving 13C-NMR showed that the supposed 7,23-derivative (based on addition to these sites in other reactions) was the 7,8-derivative (both give the same 1H-NMR pattern [121]; 7,23-bridging by the aryl group apparently involves too much strain. Both 7,8- and 7,23-addition relieve the unfavourable bond fixation that exists in the equatorial region of [70]fullerene.
10.2 Cyclopentadiene
The adduct formed by [4 + 2] cycloaddition of cyclopentadiene to [60]fullerene readily undergoes the retro Diels-Alder reaction, which can be prevented by either hydrogenation or bromination of the double bond of the addend. Uniquely, bromine adds cis instead of the normal trans, both pointing away from the cage surface (Fig. 35), attributable the unusual steric hindrance on the cage side of the double bond. Likewise the oxygen of the epoxide (produced by reaction of the cycloadduct with perbenzoic acid) also points away from the cage [122].

The product of bromine addition to 2′,3′-1′H-1,2-([1,3]epicyclopenta)[60]fullerene.
Steric hindrance also governs the reaction of pentamethylcyclopentadiene with [60]fullerene, so that the methyl group on the single carbon bridge points away from the cage [123]. Comparison of the methyl chemical shifts for the [60]- and [70]fullerene derivatives provided the first evidence that that electron withdrawal by [60]fullerene is the greater. This work also provided the first example of the differential chemical shifts between hydrogens which point towards the electronegative cage, and those which point away from it.
10.3 Anthracene
Anthracene adds readily to 6,6-bonds of fullerenes in a [4 + 2] cycloaddition, and has been used to direct further additions [124]. A unique example of addition to a 5,6-bond occurs in the reaction with C70Ph8, in which the disposition of the phenyl groups causes the 5,6-bond (C48–C49) to be a double bond, and addition takes place there in preference to any other bond [125].
Because the Diels–Alder reaction of anthracene is readily reversible, the anthracene group could be expected to migrate across fullerene cages. However, this could not be proven without a reference marker, now provided in the addition of anthracene to C60F18. Three of the possible four monoadducts were isolated, the two main isomers having Cs and C1 symmetry. The latter is the more crowded and rearranges to the former on standing, the first example of ring-walking anthracene [126].
The only known reaction of C60F20 (‘Saturnene’) occurs with anthracene giving an oxidised anthracene derivative (Fig. 36). The mechanism for this has been fully rationalised and commences with oxygen addition across the 9,10-bond of anthracene, followed by rearrangements and subsequent addition [127].

Product of the reaction of anthracene with C60F20 (‘Saturnene’).
10.4 1,3-dipolar cycloadditions
The [3 + 2] cycloaddition of C60F18 with sarcosine and formaldehyde, gave two mono- and five bis-addition products. The monoadduct obtained in the larger amount is the least hindered Cs derivative, the other having C1 symmetry, both being fully characterised. Use of benzaldehyde gave three unresolved monoadducts and two bisadducts [128]. Further derivatisation of these adducts is feasible.
10.5 Reaction of C60F18 with tetrathiafulvalene
Reaction of tetrathiafulvalene (TTF) to C60F18 takes place uniquely with concurrent [2 + 2] cycloaddition and loss of two fluorines to give a C60F16 derivative (Fig. 37). Addition of CDCl3 triggered a sterically driven elimination of thioketene (Fig. 37) observed when samples were being prepared for NMR spectroscopy. A further simultaneous unique reaction is the loss of CS2 from TTF to give a thiolactone intermediate (Fig. 38) which then adds via bond a to C60F18 with concurrent loss of 2 F; the elimination of CS2 may be catalysed by the fluorofullerene [129].
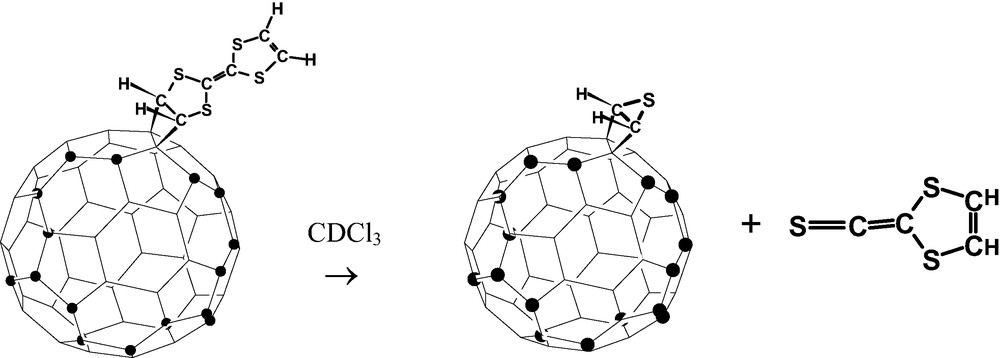
Product of the reaction of C60F18 with tetrathiafulvalene (TTF), and subsequent elimination of dithioketene (● = F).
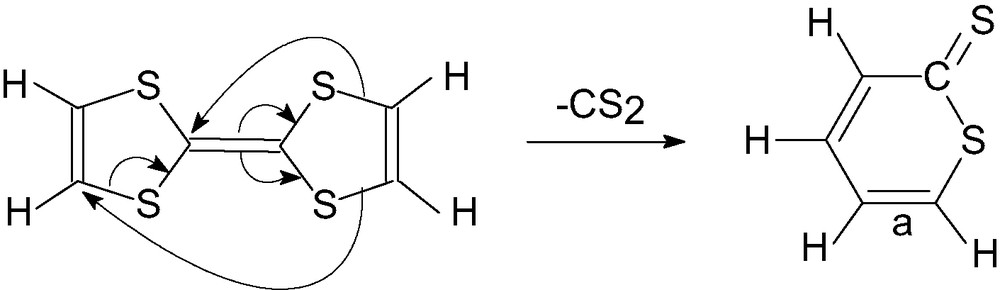
Conjectured mechanism for loss of CS2 from TTF to give a thiolactone which then adds to C60F18 with concurrent loss of 2 F.
10.6 Addition/insertion of methylene and derivatives
Methylene species, C60CH2 and C70CH2 [130], are formed during hydrogenation [15] indicating that cage degradation gives carbenes, which then either add or insert to other cages; likewise formation of CF2 adducts accompanies fluorination [39].
Polymethylene adducts, e.g. C60(CH2)1-6 and C70(CH2)1,2 [130,131], are obtained in particular on heating fullerenes or bromofullerenes with THF, the trimethylene derivatives dominating [132]. These appear to be propano compounds since they lose 2H during mass spectrometry (not possible if separate methylene groups were involved). The mechanism may involve elimination of formaldehyde from THF, since the corresponding reaction with dihydropyran gives preferential formation of derivatives containing four methylene groups.
Methanofullerenes C60(CHCN) and C70(CBr2), obtained by reaction of [60]fullerene with either CH2BrCN or CHBr3 in the presence of LDA, have synthetic potential yet to be explored [133].
11 η6 co-ordination to a fullerene
The ability to co-ordinate a transition metal to a fullerene has long been sought, but has been denied by the lack of aromaticity, so co-ordination with fullerenes has been limited to η2 (typical of an alkene) [134] or η5 (to a cyclopentadienide ring) [135].
The first example of η6 co-ordination has now been obtained through reaction of C60F18 (which has a fully aromatic planar ring) with [Mo(CH3CN)3(CO)3] whereby displacement of acetonitrile gives the product shown in Fig. 39. It was not possible to obtain a single-crystal X-ray structure, but the characterisation was accomplished by a very detailed mass spectrum; the compound is quite stable to the extent that a parent ion at 1244 amu was readily obtainable [136].
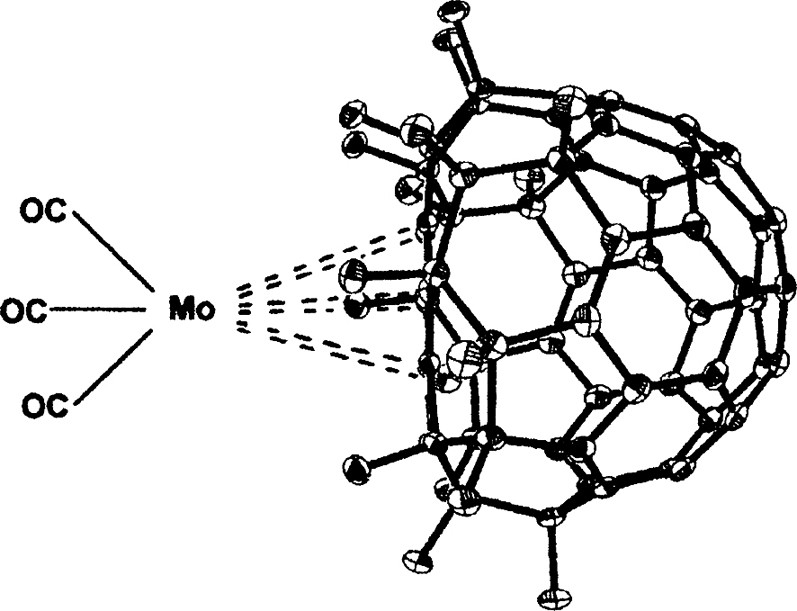
Structure of [MoC60F18(CO)3].
12 Conclusion
The present paper summarizes the work accomplished in our group on the chemical modification of fullerenes. Fluorine addition to C60 can be performed with a variety of agents. The difficulty of controlling the reaction provides a complex mixture of products with varying numbers of fluorine atoms attached to the carbon cage and inhibits the isolation of the early compounds of fluorine addition. Nonetheless, many fluorofullerene derivatives have been characterized over the years. In general, fluorine addition to fullerenes presents two main features: the fluorine atoms are attached to the cage as fluorine pairs across 6,6-bonds, and the fluorination has a general tendency to create structures with increased aromaticity relative to the fullerene precursor and hence with increased stability. In other words, the aromatic character of the fluorofullerenes increases with increasing number of fluorine atoms attached to the cage. Because fullerenes are moderately aromatic, addition of the first fluorine pair increases the localization of the electrons for the remaining double bonds of the two hexagons involved, so that further addition to these hexagons becomes thermodynamically favoured. Chlorination and bromination reactions have also been studied. These radical reactions have steric requirements different from fluorination resulting in quite different regiochemistry, with 1,4-addition being a dominant feature. Other investigations of the chemistry of fullerenes have been concerned with their use as electrophiles for aromatic substitution, initial reactions being carried out using unhalogenated [60]fullerene in a Friede-Crafts reaction. Other studies involved the use of the more electrophilic halogenofullerenes. These can be generated in situ, for example, using a mixture of aromatic compounds, bromine, ferric chloride and [60]fullerene, but like the foregoing reactions, gave products that were difficult to separate and/or characterise. The most successful studies have used reactions of preformed chlorofullerenes with aromatic compounds (mostly benzene), which proceed via intermediate formation of fullerene carbocations. The use of a fluorofullerene as an electrophile has also been reported involving the FeCl3-catalysed reaction of C60F18 with benzene, which produced the triphenyl derivative (‘triumphene’) (1, Ar = Ph). In conclusion, our studies have clearly shown how fullerene chemistry is governed by a combination of steric effects, electron withdrawal by the cages, a drive to increase their aromaticity, and an increase in localization of π-electrons following a first addition.