

1 General considerations
Photoinduced energy and electron-transfer reactions are the key steps in natural photosynthesis, and the elucidation of their mechanisms continues to attract considerable interest [1–6]. Similar processes occur in artificial photoactive and telectroactive molecular donor–acceptor systems [7–10]. Such systems are considered to be promising for applications in molecular and supramolecular electronics, light harvesting, and photocatalysis [11,12]. Molecular donor–acceptor arrays also find application in organic and polymer photovoltaic cells that convert light into electric energy [13]. A blend of a chromophore that can harvest sunlight in a very efficient way as a donor and a fullerene derivative as the acceptor rank among the most promising materials for photovoltaic cells [14–18]. These hybrids contain C60 bulk heterojunctions [19], so that the forward electron transfer (ET) is extremely fast (in the picosecond range), whereas back electron transfer (BET, charge recombination) extends to the millisecond time domain or even longer. According to Marcus theory [20], the large driving force of the BET process in charge-separated (CS) states generated from hybrids in which fullerene acts as the electron acceptor, coupled with a low reorganization energy for such systems, pushes BET into the inverted region of the Marcus curve, sharply reducing the rate of this process. The large difference between the forward and backward ET rates, ranging up to four orders of magnitude in systems studied to date [21,22], ensures rapid and efficient generation of CS states, together with the opportunity to transport and collect the photogenerated charges.
For example, Imahori et al. [23] synthesized multicomponent porphyrin–fullerene systems that exhibit charge-separated state lifetimes of on the order of microseconds. Later, they bound a similar porphyrin–fullerene dyad to a gold surface using a sulfide bridge, and observed photocurrents from this construct incorporated into a photoelectrochemical cell [24].
Efforts have been directed mainly to preparation of covalently linked donor–acceptor systems as photosynthetic models, in attempts to efficiently generate long-lived charge separated (CS) states [25–27]. Particular attention has been given to systems in which free base and metalloporphyrins [28], ferrocene [29], and metal complexes [30,31] such as ruthenium tris-bipyridyl ([Ru(bpy)3]2+, act as the electron donors, and [60]fullerene (C60) is the ultimate electron acceptor, sometimes in multicomponent arrays. The topology of these systems is controllable by the nature of the linker(s), which can be either flexible or rigid. For covalently-linked porphyrin–fullerene (P–F) hybrids, two extremely important general characteristics are generally observed: (1) if the linker has sufficient conformational flexibility, the P and F moieties tend to be in close spatial proximity because of strong van der Waals attractive forces (π–π interactions), which enhances intramolecular electronic interactions, e.g. kcs, the rate of photoinduced charge separation, can be 10 ps–1 or less; (2) the rate of charge recombination (back electron transfer, BET) kcr is generally much less than kcs, often by several orders of magnitude, for the reasons discussed above. Electron transfer almost always dominates over competitive singlet–singlet energy transduction (EnT) for multicomponent hybrids constructed from free base porphyrins in relatively polar solvents—benzonitrile, tetrahydrofuran, dimethylformamide—as well as for metalloporphyrins (ZnP) in both nonpolar—benzene, toluene—as well as polar solvents—tetrahydrofuran, benzonitrile, dimethylformamide.
2 Porphyrins and fullerenes in supramolecular systems
The design and synthesis of light-harvesting systems that rival those in photosynthesis requires the organization of several components in an elaborate molecular topology. Porphyrins are the most frequently employed electron donors in photosynthetic model systems because they are excellent light-harvesting antennas and exhibit well-known photophysical properties. For the reasons discussed above, increasing attention has been given to incorporation of C60 as the electron acceptor in photosynthetic model systems. Thus, many types of porphyrin–C60 hybrids with flexible as well as rigid covalent linkages have been prepared for study of their ET properties.
However, in the natural photosynthetic reaction centers, the chromophores are embedded in a protein matrix and are not covalently linked together [6]. The interactions between the electron donor and acceptor components are highly complicated as well as extremely efficient. Thus, study of the PET process in porphyrin–fullerene supramolecular systems, in which the electron donor and acceptor are mechanically rather than covalently linked, should enhance our understanding of the natural system.
Although fullerenes are recognized to be superior electron acceptors with regard to their redox potential, reorganization energy, and structure, only a limited number of noncovalently linked model systems have been constructed to address some of the most basic questions regarding the effect of distance and orientation on the rates of electron transfer [32–34].
Rotaxanes possess a macrocyclic ring which is threaded on a rod with stoppers that prevent dissociation, while Catenanes possess two (or more) non-bonded interlocking rings. These nanoscale systems are distinct from pseudorotaxanes, which can undergo dissociation into their individual components [35]. Since rotaxanes and catenanes are interlocked species, dissociation requires breaking of a covalent bond. However, the boundary between rotaxanes and pseudorotaxanes is not always clear. For example, when the stoppers are not large compared to the hole in the macrocyclic component, a rotaxane at low temperature might well become a pseudorotaxane at higher temperature. An example of such a system will be discussed later.
Sauvage and coworkers have pioneered the study of long-range ET processes in purely porphyrinic D–A systems with rotaxane and catenane topologies, in which the D and A moieties are mechanically linked [28,36,37]. A general and highly effective strategy to prepare such rotaxanes and catenanes in high yields is based on the metal template effect, which relies on the presence of molecular recognition sites between the components to be assembled. In the metallocatenane and rotaxanes constructed by Sauvage and coworkers, the coordinating rings contain 2,9-di-p-phenyl-1,10-phenanthroline (phen) moieties, which, upon complexation to Cu(I), adopt an entwined topography. The highly rigid system thus obtained favors distorted tetrahedral geometries around Cu(I). Sauvage et al. studied long range photoinduced ET processes in rotaxanes and catenane architectures incorporating electron donor (ZnP) and electron acceptor (Au(III)P) moieties, such as
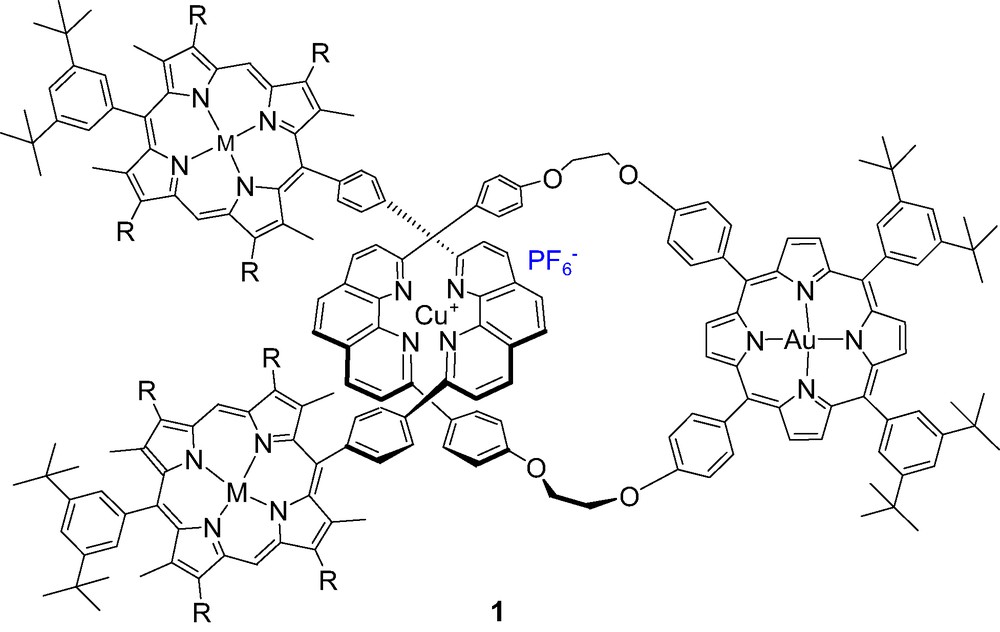
compound 1 [38]. In this rotaxane, where the ZnP and AuP moieties are separated by 15–17 Å edge-to-edge, PET is very fast [28]. Biexponential decay of ZnP singlet excited states is observed, 70% involving ET to AuP+ (60 ps) and 30% involving EnT to Cu(phen)2+ (300 ps) followed by slower (650 ps) generation of ZnP triplets (3ZnP*). Decay of the CS state is biexponential with lifetimes of 10–40 ns, which is attributed to different conformations that exhibit different degrees of electron coupling. Selective excitation of AuP+ results in enhanced ET mediated by the Cu+ moiety, giving ultimately the same CS state as obtained on excitation of ZnP. However, from the point of view of energy storage, Au(III)P+ is not a good electron acceptor, because the rate of back electron transfer is much too fast, ranging from the ps to the short ns time scale, depending on the precise topology of the system.
The Cu(I) ion can be removed in Sauvage's systems by treatment with excess KCN, which changes the topology of the system by eliminating the tight tetrahedral coordination. This allows slippage of the thread or the rings through the macrocycle, accessing conformations not possible in the presence of the Cu(I) complex, as illustrated in Fig. 1. Sauvage has shown that this completely changes the dynamics of forward and back ET processes.
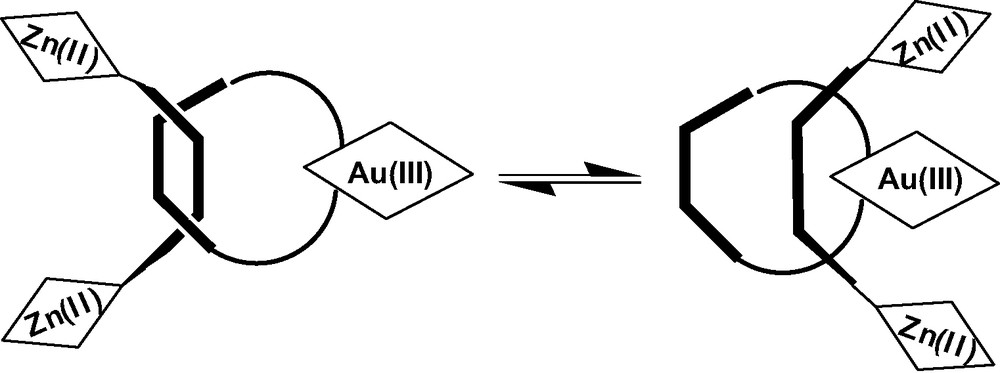
Sauvage's demetallated rotaxane.
Because C60 is an excellent electron acceptor, and has a small reorganization energy which retards the BET process, we postulated that changing the electron acceptor in Sauvage-type rotaxanes and catenanes to C60, should lead to much longer lived CS states, thus extending the applicability and utility of such nanoscale systems. As will be discussed below, our research in this area, inspired by that of Sauvage, involves synthetic modifications by which C60 moieties can be incorporated on either the macrocycle or the thread prior to or following assembly of rotaxane and catenane architectures.
3 Previously reported rotaxanes incorporating fullerenes
The first reported rotaxane of this type was a Cu(I)phen2 complex 2 in which the thread had two C60 stoppers [39,40]. In this structure, silylmethanofullerene moieties were attached to phen groups by rigid bis-alkyne linkers, and the supramolecular structure was then assembled using Sauvage's Cu(I) template strategy. Excitation of the fullerene-stoppered rotaxane at 355 and 532 nm leads to partitioning of absorption between the Si–C60 and Cu-(phen)2 moieties, based on spectra of model compounds. It was proposed that energy transfer (EnT) from the fullerene-centered S1 state to the Cu(I)- phen2 (k = 1.6 × 109 s–1) dominates over ET, since there was no spectroscopic evidence for formation of the CS state on this timescale, even though ET has a larger driving force (ΔG0 = 0.27 eV for ET vs. 0.08 eV for EnT). The EnT process was argued to take place by an electron exchange rather than a Förster mechanism. Intersystem crossing to C60 triplets is a minor decay process. In turn, the excited Cu(I)-complex is quenched
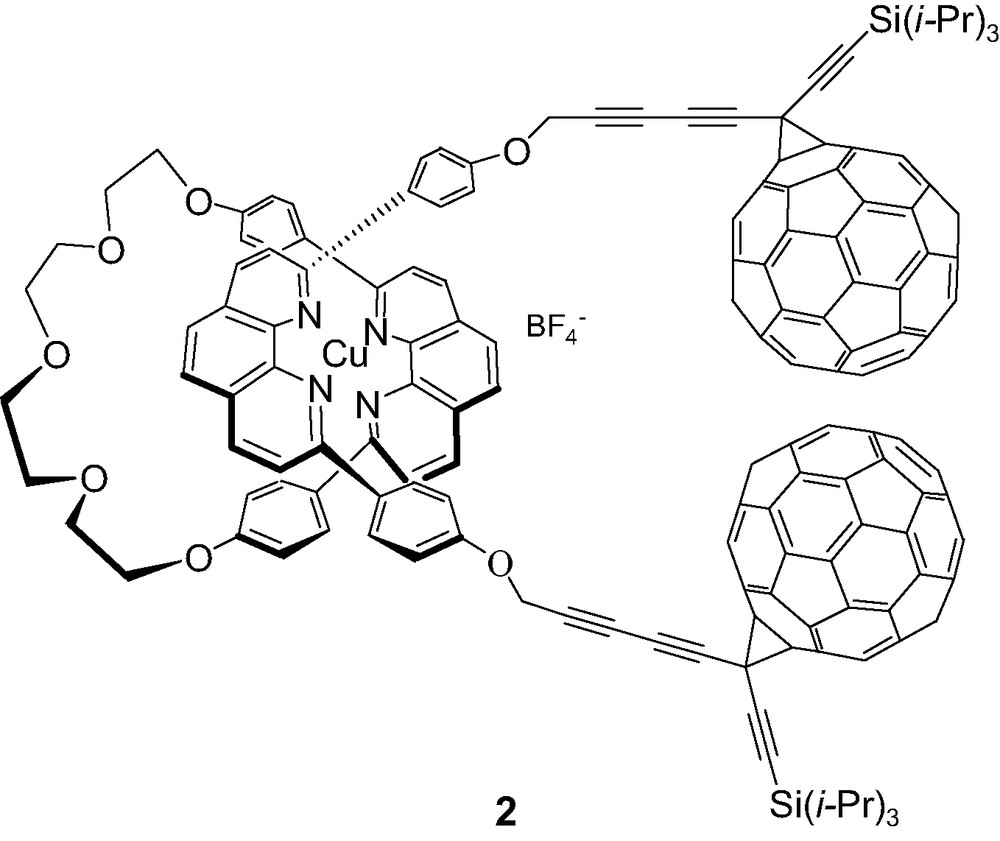
mainly by ET to form the CS state, i.e., the oxidized Cu(I)phen2 complex and the C60 radical anion, in competition with slower formation of C60 triplets. These triplets are also quenched (τ = 170 ns vs. 5 μs for a Si–C60 model compound), presumably by formation of the CS state, a pathway that is slightly exergonic (ΔG0 ~ 0.03 eV). However, no clear-cut evidence for formation of the CS state was obtained from transient absorption spectra. A weakly absorbing (~ 740 nm) species with a lifetime of 1.7 μs was observed, which the authors are reluctant to assign to the CS state because of its unusually long lifetime. Due to instrumental limitations observations were not made in the region above 850 nm where the C60 radical anions absorb (~ 1000 nm). These authors concluded that BET was too fast in this system to allow observation of the CS state. Based on the work described below, we believe that it is more likely that the long-lived transient produced from this rotaxane is indeed the sought-after CS state.
A second example of a porphyrin–fullerene rotaxane was reported by Ito's group (Watanabe et al [41] and Sandanayaka et al. [42]). In 3, the fullerene is appended to the macrocycle, with two tetraarylporphyrin stoppers on the thread. This rotaxane was synthesized in two steps: spontaneous assembly of the basic rotaxane skeleton from its components by a hydrogen-bond assisted strategy, followed by attachment of the fullerene. The absorption spectrum is essentially a superimposition of the spectra of C60 and the bis-porphyrin rotaxane precursor. The ZnP fluorescence is strongly quenched relative to that of the precursor lacking the C60. There are two components to the fluorescence decay curve, 93 ps (80%) and 1.6 ns (20%), the latter only slightly longer than that of the precursor lacking the fullerene.

The transient absorption spectrum taken 100 ns after the flash shows characteristics of both ZnP radical cations (850 nm) and C60 radical anions (1000 nm); the latter absorption could be clearly detected 6 ns after the pulse, indicating that the major component of the ZnP fluorescence decay is ET to give the CS state. The quantum yield and rate constants for charge separation, Φcs and kcs, were 0.95 and 1.0 × 1010 s–1, respectively. The lifetime of the CS state, derived from the decay of the absorption at 1000 nm, is 180 ns in benzonitrile. Based on estimates of the rates of through-space and through-bond ET, it is concluded that BET takes place through space by a superexchange mechanism. Due to its structure, there is considerable flexibility in the relative orientation of the ZnP and C60 chromophores in 3, which is reflected in the dynamics.
4 Rationale
Based on the previous work in this field described above, we concluded that incorporating fullerenes into Sauvage-type rotaxanes and catenanes should greatly further our understanding of the relationship between molecular topology and rates of forward and backward electron transfer. Due to the differences of the reorganization energies of fullerene compared to the traditional quinone and the Au(III)P acceptors, the shift of the Marcus curve should place the BET process in fullerenoporphyrinorotaxanes and catenanes deep into the inverted region [17]. As a result, the BET process in these novel fullereno D–A architectures should be much slower compared to the CS process. Photophysical studies, in collaboration with eminent scientists in this field, fully substantiated this hypothesis.
5 Porphyrin-stoppered fullerorotaxanes
Our initial efforts were focused on the preparation of the bis-porphyrin stoppered rotaxane systems shown in Fig. 2 [43]. Rotaxane 4 lacking the C60 moiety served as a model comparison for photophysical comparison with the fullerene-linked rotaxanes 5, 6 and 7. Because of the tetrahedral arrangement of the phen ligands around Cu(I), the electron donor (P) and electron acceptor (F) are separated by ~2 nm, which prevents both CS and CR processes from occurring directly through space.
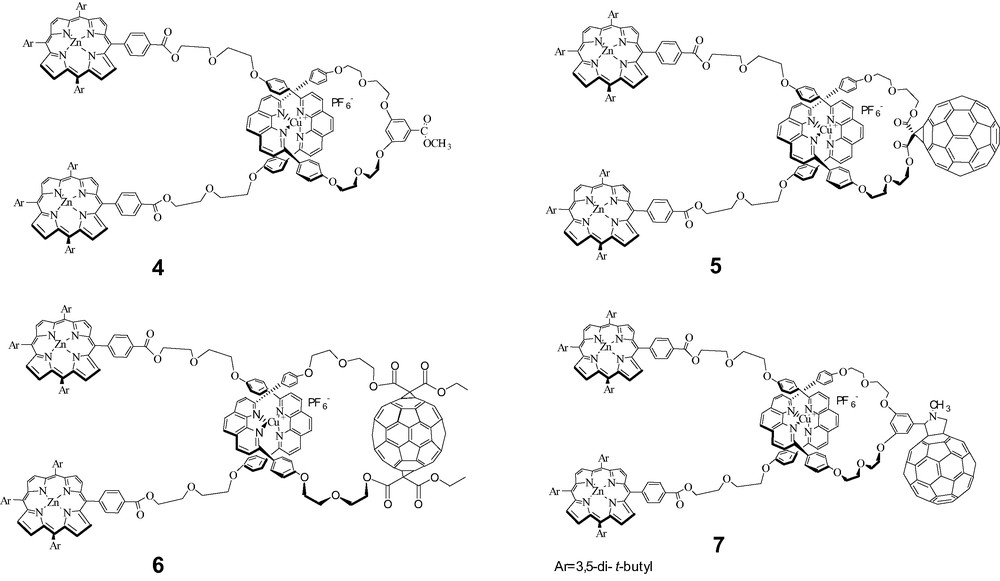
Target porphyrin-stoppered rotaxanes.
The general synthetic approach is shown schematically in Scheme 1. First, one needs to synthesize the phen macrocycle corresponding to 4 and the fullerenomacrocycles corresponding to structures 5, 6 and 7, by Bingel [44] and Prato (Maggini et al. [45]) reactions, respectively. Treatment of the phen-containing macrocycles with Cu+(MeCN)4PF6– in MeCN/CH2Cl2 leads to formation of the tetrahedral Cu(I) complex, which is stabilized by MeCN in the mixed solvent system. The linear phen thread with a zinc tetrarylporphyrin stopper at one end is then added, forming a new tetrahedral Cu(I) complex by expulsion of the MeCN ligands on copper. The synthesis is completed by EDC/DMAP coupling to a second ZnP group at the other end of the thread.
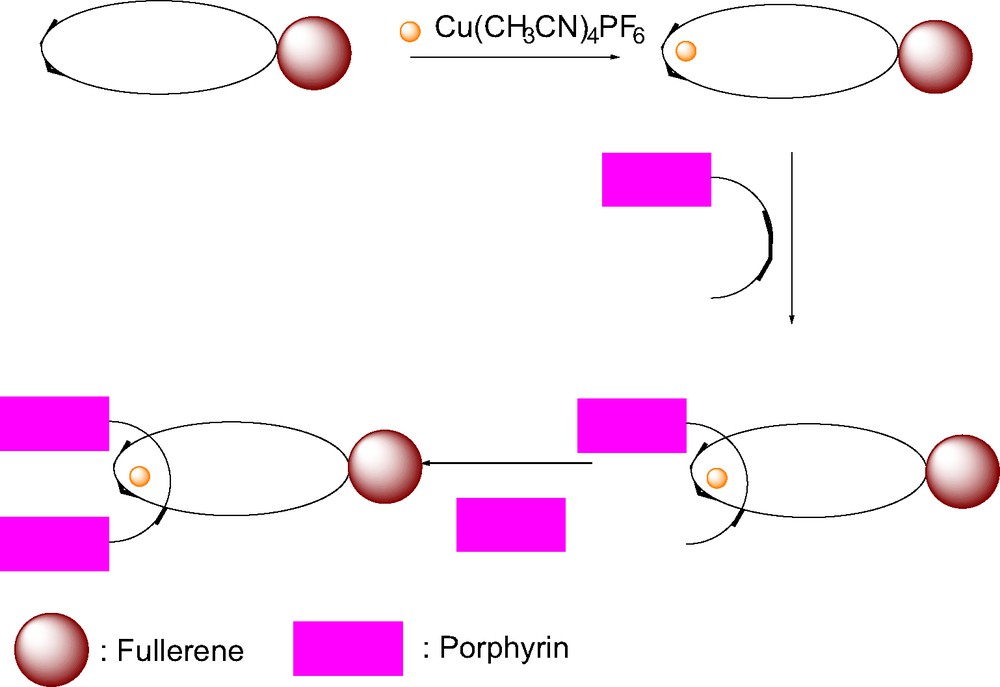
Synthetic approach to porphyrin-stoppered rotaxanes.
The entire synthesis leading to rotaxane 5 is shown in Scheme 2, starting with phen synthon 8 originally reported by Sauvage [46]. In this synthesis, BOP-Cl/Et3N was found to be the best coupling reagent for macrocyclization to give the macrocyclic malonate 9 (~30% yield). The malonate was smoothly transformed to the fulleromacrocycle 10 via the Bingel–Hirsch reaction in reasonable yield by using an excess amount of C60. The 1H-NMR spectrum of 10 is distinguished from its precursor by (a) the disappearance of the singlet at δ 3.45 ppm for the two acidic protons on the malonate, and (b) significant downfield chemical shift for the two pairs of ester protons from δ 4.2 to 4.5 ppm due to the influence of the attached C60. The structure of 10 was confirmed by MALDI-TOF and high-resolution FAB-MS data and 1H-NMR.
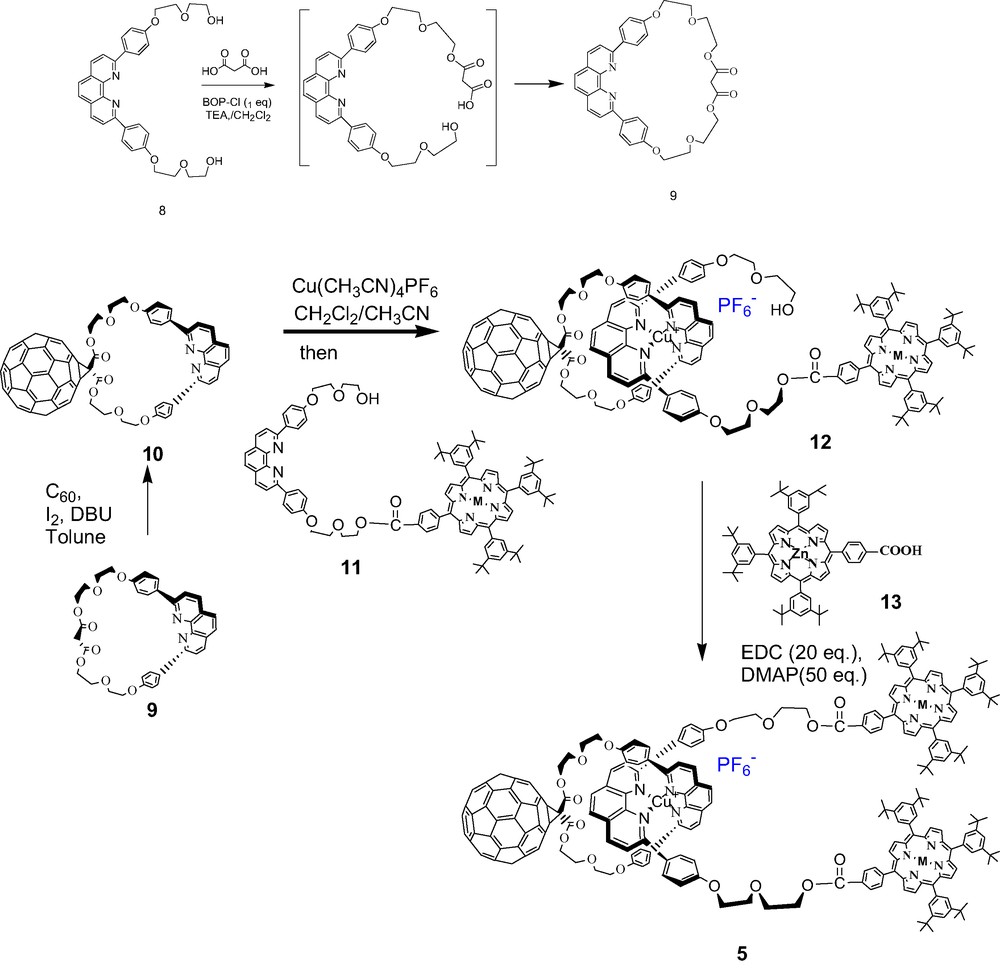
Synthesis of rotaxane 5.
The mono-ZnP-stoppered linear thread 11 was synthesized from tetraarylporphyrin carboxylic acid and 8 via EDC/DMAP coupling, and then metallated with Zn(OAc)2. Phen thread 11 was added to a solution of 10 in degassed dry CH2Cl2/CH3CN (3:2) to give the stable tetrahedral Cu(I)phen2 complex 12. The process is easily monitored by TLC by the disappearance of 10 and of the fluorescence of 11. A new spot on the TLC plate had the characteristic dark purple color of a porphyrin, but was not fluorescent under UV irradiation. The fluorescence quenching process in 12 and related materials will be discussed later.
The 1H-NMR spectrum of linear thread 11 provides valuable information with respect to the synthesis. Although Amabilino and Sauvage reported that one of their porphyrinomacrocycles shows concentration dependent behavior due to π–π stacking [47], it was quite surprising to see this for 11, which has considerable conformational flexibility. In the aromatic region, two sets of the signals are seen for the phenanthroline protons (one set of aromatic protons show a pronounced upfield shift) due to the loss of symmetry compared to its precursor 8, consistent with the unsymmetrical structure for 11. The methylene protons on the diethylene glycol linker are also much better resolved in 11 than in 8 and seven signals are observed for the eight types of methylene protons, some of which are shifted significantly upfield due to the porphyrin moiety. The complexation reaction is much slower with 11 than with 8 because (a) there is only one direction for 11 to thread through the macrocycle, and (b) the thread needs to convert from a π–π stacked conformation into a fully extended conformation for threading to occur.
The syntheses of the other ZnP-stoppered rotaxanes (4, 6 and 7) were carried out in an analogous manner to that shown in Scheme 2. For synthesis of 7, the ester group in the macrocyclic precursor to 4 was subjected to a reduction-oxidation sequence (LiAlH4 in THF, followed by MnO2 in CHCl3) to give the corresponding aldehyde. Reaction of the macrocyclic aldehyde and N-methylglycine led in situ to formation of an azomethine ylide which underwent 1,3-dipolar cycloaddition to C60 to give the macrocyclic fulleropyrrolidine precursor to 7. The structures of all synthetic intermediates and the final rotaxanes were fully supported by mass, UV-Vis and 1H-NMR spectral data.
6 Catenanes derived from porphyrin-stoppered rotaxanes
Molecular modeling of porphyrin-stoppered fullerenorotaxane 5 and model rotaxane 4 suggested that the two ZnPs linked to the Cu(I)phen2 core constitute a molecular scaffold that should exhibit excellent binding features toward a variety of bidentate guest molecules that coordinate to ZnP, as shown schematically in Scheme 3.
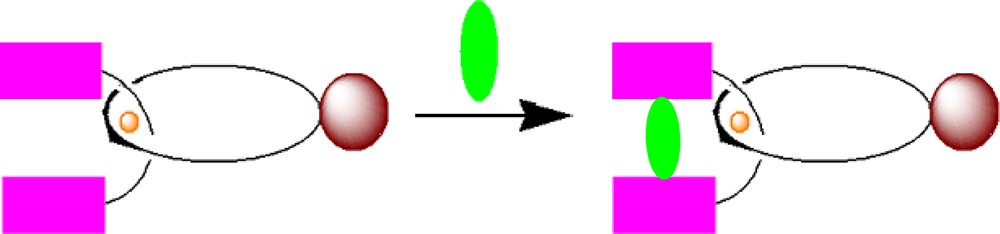
Conversion of rotaxane 2 into catenanes by complexation with bidentate ligands.
Thus, selective complexation of the free nitrogen lone pairs of DABCO or 4,4′-bipyridyl to the axial dz2 orbitals of ZnP opens pathways to novel catenanic architectures shown schematically in Schemes 4 and 5 [47].
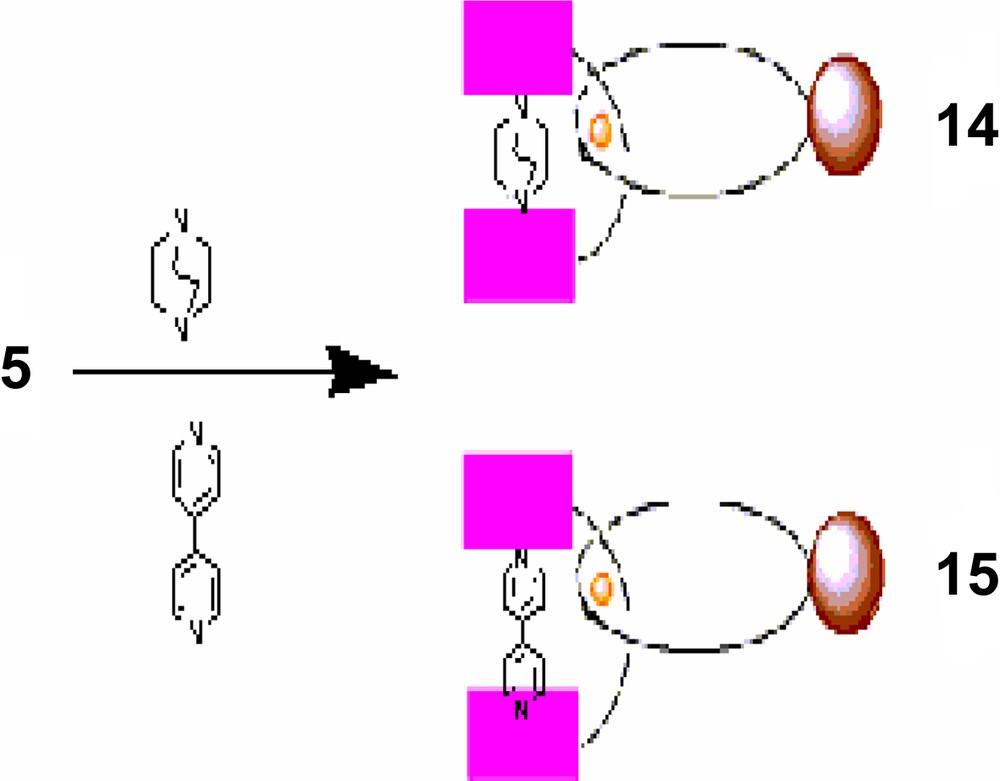
Formation of catenanes from 5 using DABCO and 1,4-bipyridyl.
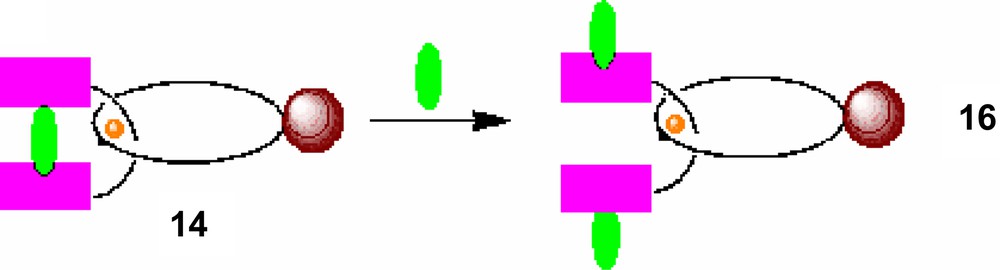
Transformation of catenanes to rotaxanes using excess DABCO.
UV-Vis and NMR spectra were employed to follow binding of DABCO and 4,4′ bipyridyl) to the bis-ZnP scaffold in rotaxanes 4 and 5 as described previously [48]. A typical titration experiment is illustrated in Fig. 3 for the addition of 4,4′-bipyridyl (in concentrations ranging from 4.6 × 10–7 to 1.5 × 10–5 M) to rotaxane 4 (1.0 × 10–6 M in CH2Cl2). The isosbestic point at 425 nm testifies to a clean rotaxane–catenane transformation. The ground state absorption shifts were analyzed within the framework of the Benesi–Hildebrand equation, leading to association constants Ka of 2.0 ± 0.5 × 105 M–1 and 1.8 ± 0.5 × 105 M–1 for complexation of rotaxanes 4 and 5 with DABCO and 4,4′- bipyridyl, respectively. Fig. 4 depicts the corresponding changes in fluorescence observed on addition of bipyridyl to rotaxane 5. The complexation in all cases can be reversed simply by the addition of dilute HCl solution, which protonates the nitrogen lone pair electrons [48].
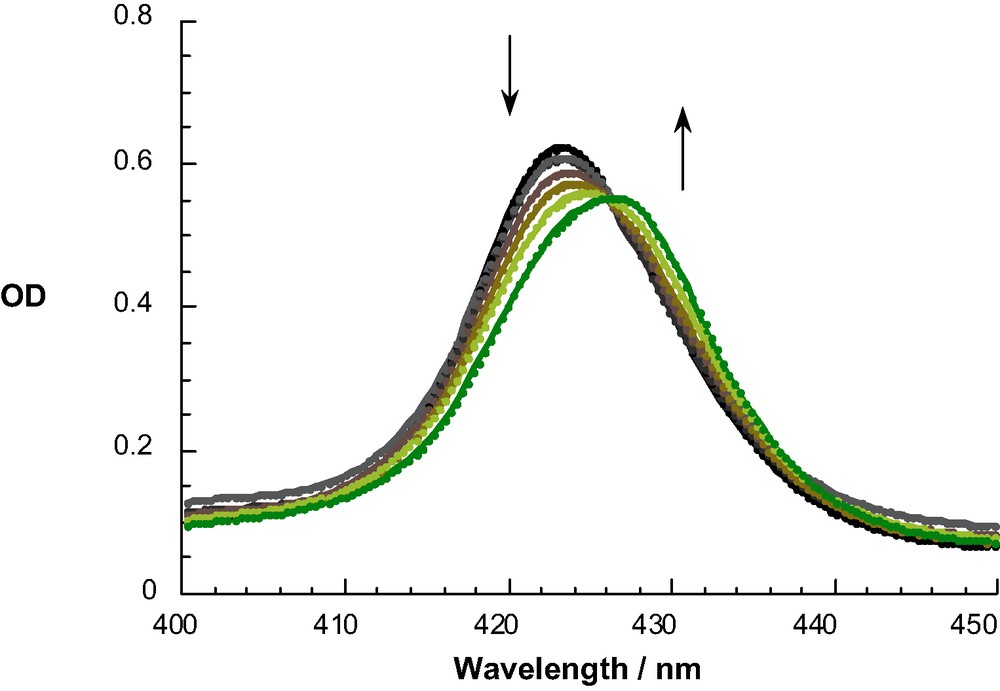
Absorption spectra of rotaxane 5 (0.9 × 10–6 M) in dichloromethane with various equivalents of 4,4′-bipyridyl (4.6 × 10–7 M–1.5 × 10–5 M) to form catenane 16.
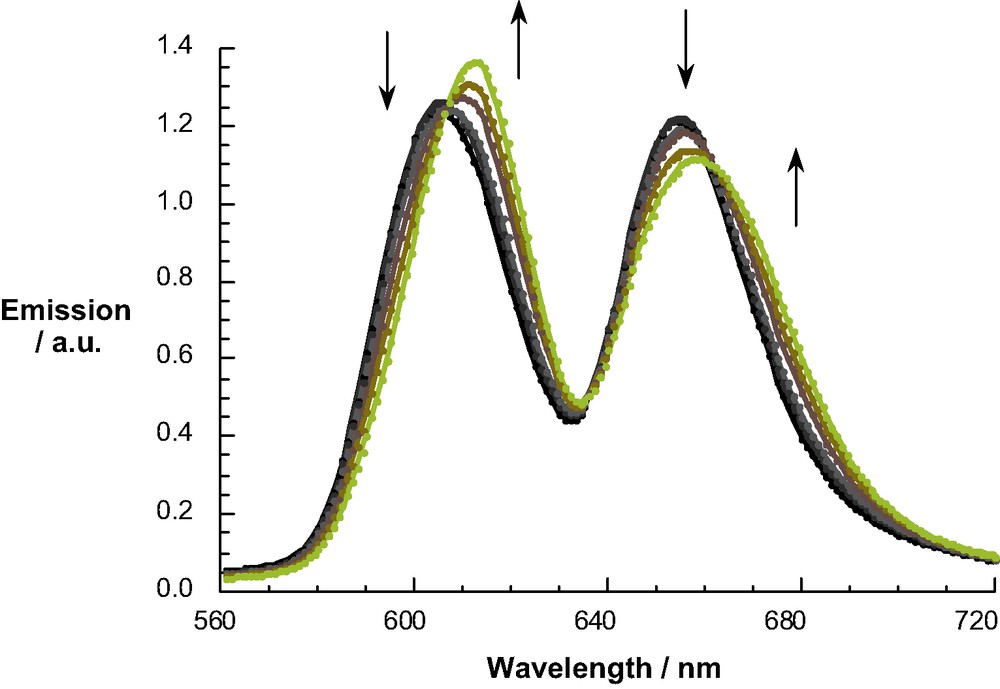
Fluorescence spectra of rotaxane 5 (0.9 × 10–6 M) in dichloromethane with various equivalents of 4,4′ bipyridyl (4.6 × 10–7 M–1.5 × 10–5 M) to form catenane 16; the excitation wavelength was 425 nm, the isosbestic point observed in the absorption spectra (see Fig. 3).
7 Fullerene-stoppered porphyrinorotaxanes [49]
In the second set of compounds, the location of the ZnP and C60 moieties is reversed from that described above, i.e. the ZnP is appended to the macrocycle and the fullerenes are the stoppers on the thread around the central Cu(phen)2+ core. As will be seen, this change in topology results in enhanced lifetimes of the charge-separated radical pair (CSRP) state produced on photoexcitation of these materials.
Our first attempt at synthesis of these materials involved adaptation of the route used to prepare rotaxanes 4, 6, and 7 described above, as illustrated schematically in Scheme 6. In this approach, a phen thread with a fullerene stopper is passed through a symmetrical phen-containing macrocycle attached to a tetraaryl ZnP moiety, followed by attachment of a second fullerene stopper. For this purpose, compound 18 was prepared as shown in Scheme 7. However, this material was not very soluble in the CH2Cl2/CH3CN solvent system required for the Cu(I)-templated rotaxane synthesis, and also tended to aggregate in this medium. This approach was therefore abandoned.
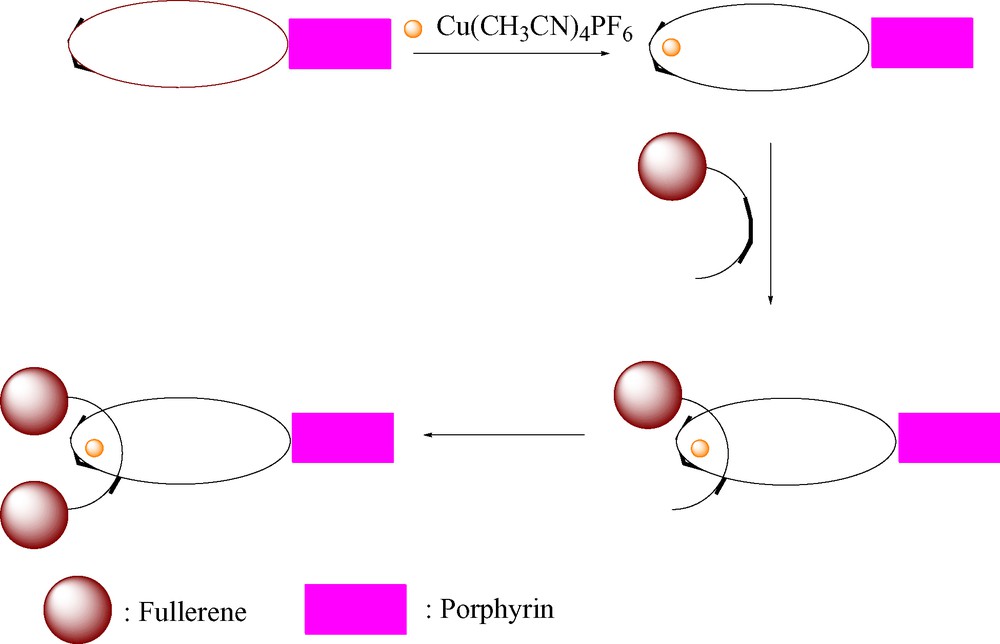
Schematic synthesis of fullerene-stoppered porphyrinorotaxanes.
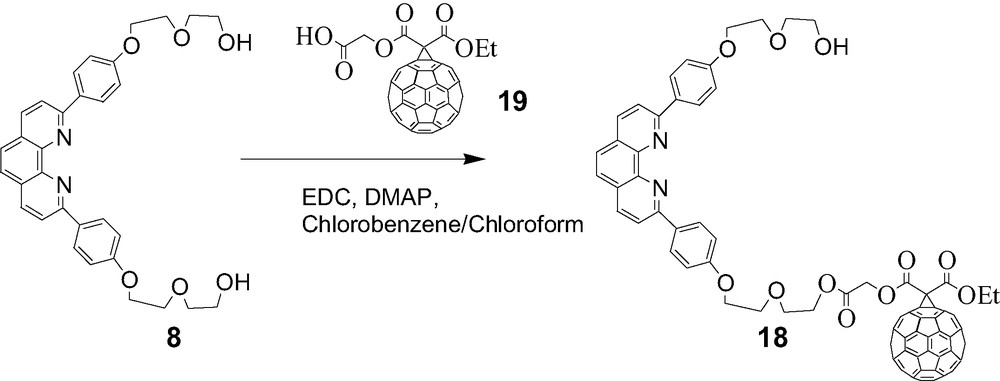
Synthesis of compound 18 from 8.
A second approach, illustrated schematically in Scheme 8, involves insertion of an unstoppered phen thread through the macrocycle followed by attachment of two fullerene stoppers. Using this approach, three fullerene-stoppered rotaxanes 20, 21 and 22 were prepared. The specific routes to each of these compounds are shown below.
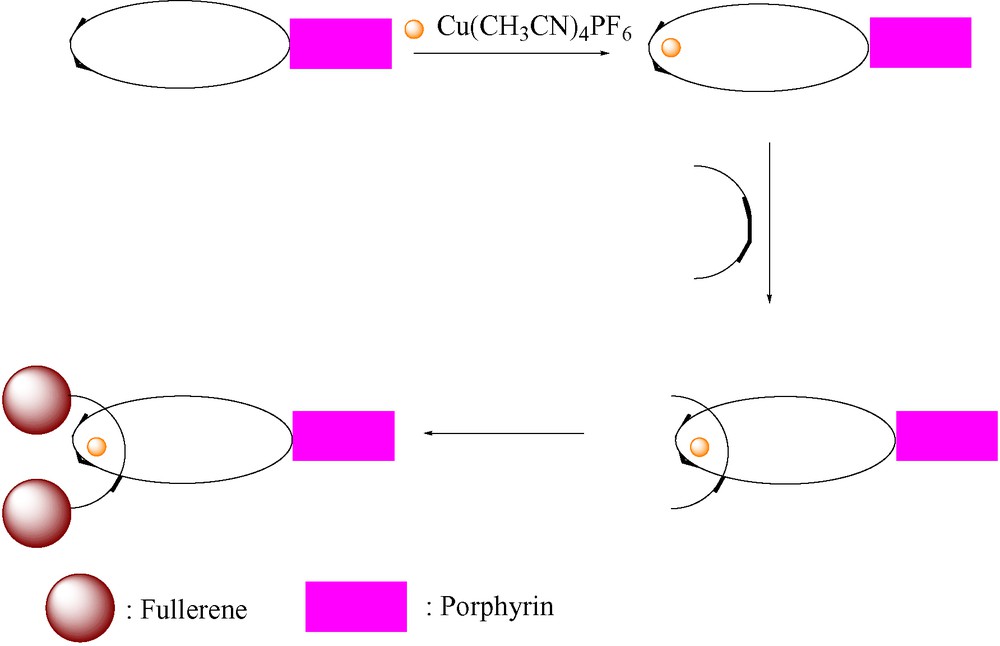
Alternative approach for synthesis of fullerene-stoppered rotaxanes.
Macrocycles were designed to which a porphyrin was either incorporated or appended using standard coupling reactions. Porphyrinomacrocycle 23 was prepared according to the literature. Complexation with Cu(I) was monitored by TLC and MALDI-TOF mass spectrometry. Formation of the tetrahedral Cu(I) complex using the unstoppered linear phen-containing thread 8 was carried out as per Sauvage. The final step involved an activated coupling reaction with the C60 carboxylic acid synthon 19 at room temperature. Aminotetraphenylporphyrin 24 required for the synthesis of rotaxane 22 was synthesized according to a literature procedure.
All new materials were fully characterized by mass, 1H-NMR and UV-Vis spectra (Schemes 9–11).
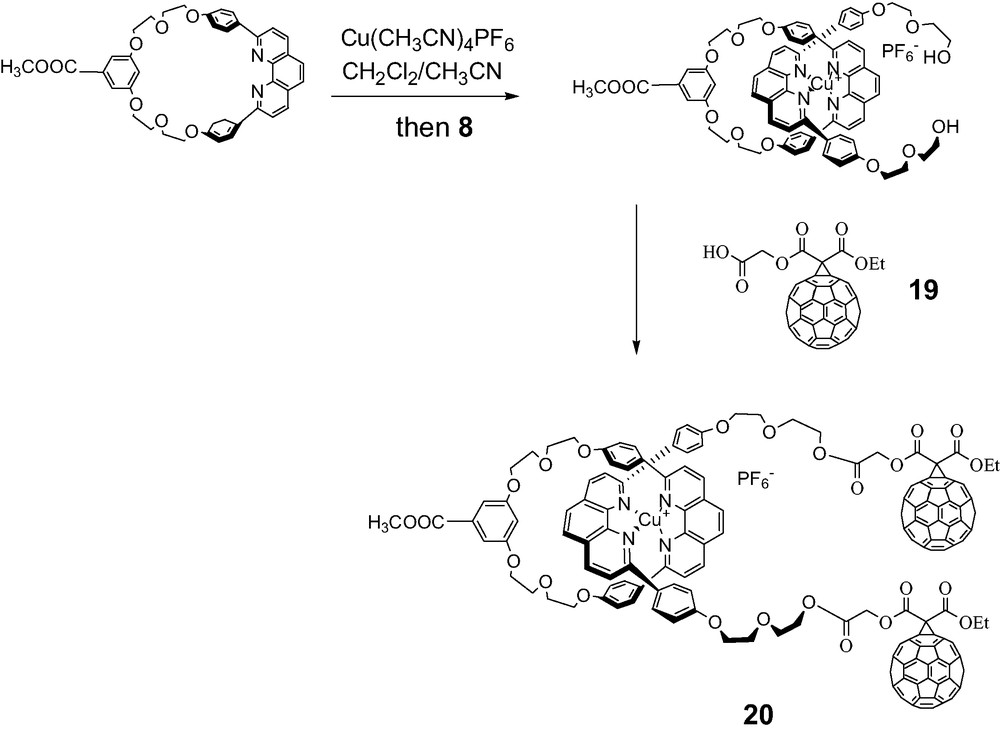
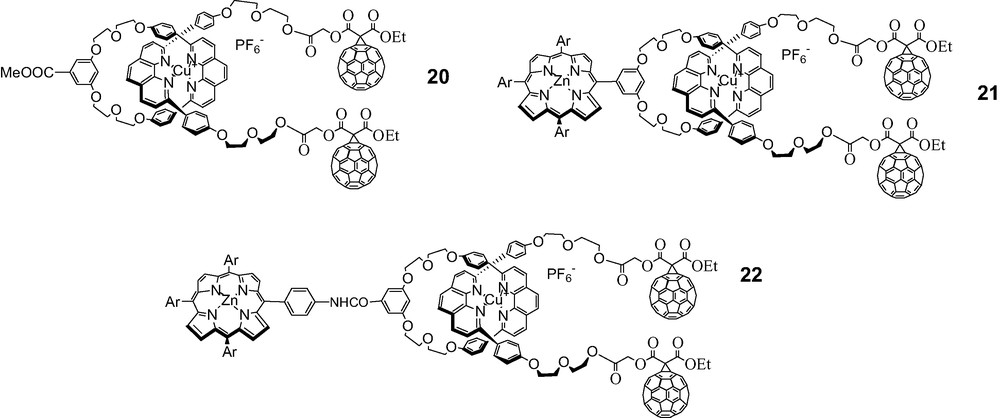
Synthesis of fullerene-stoppered rotaxane 20.
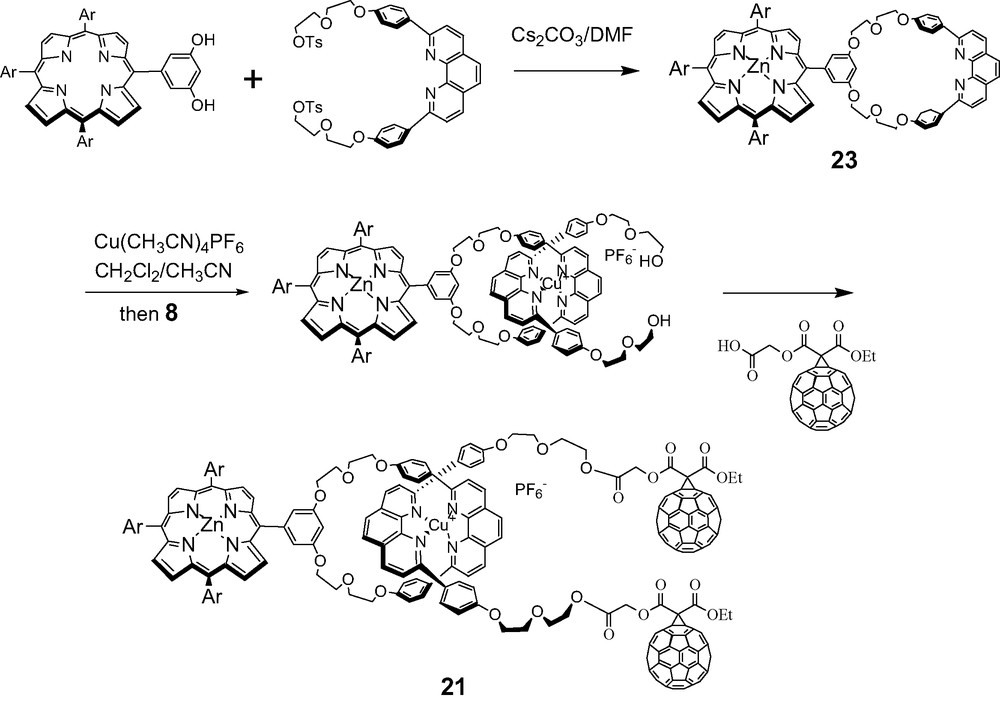
Synthesis of fullerene-stoppered rotaxane 21.
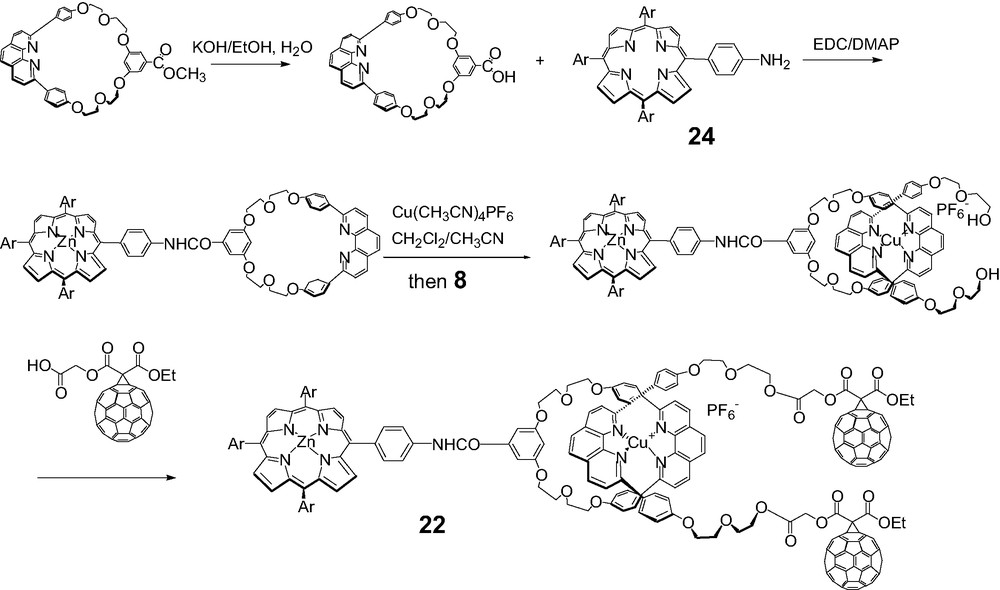
Synthesis of fullerene rotaxane 22.
8 Transition-metal-assisted self-assembly of fullerorotaxanes
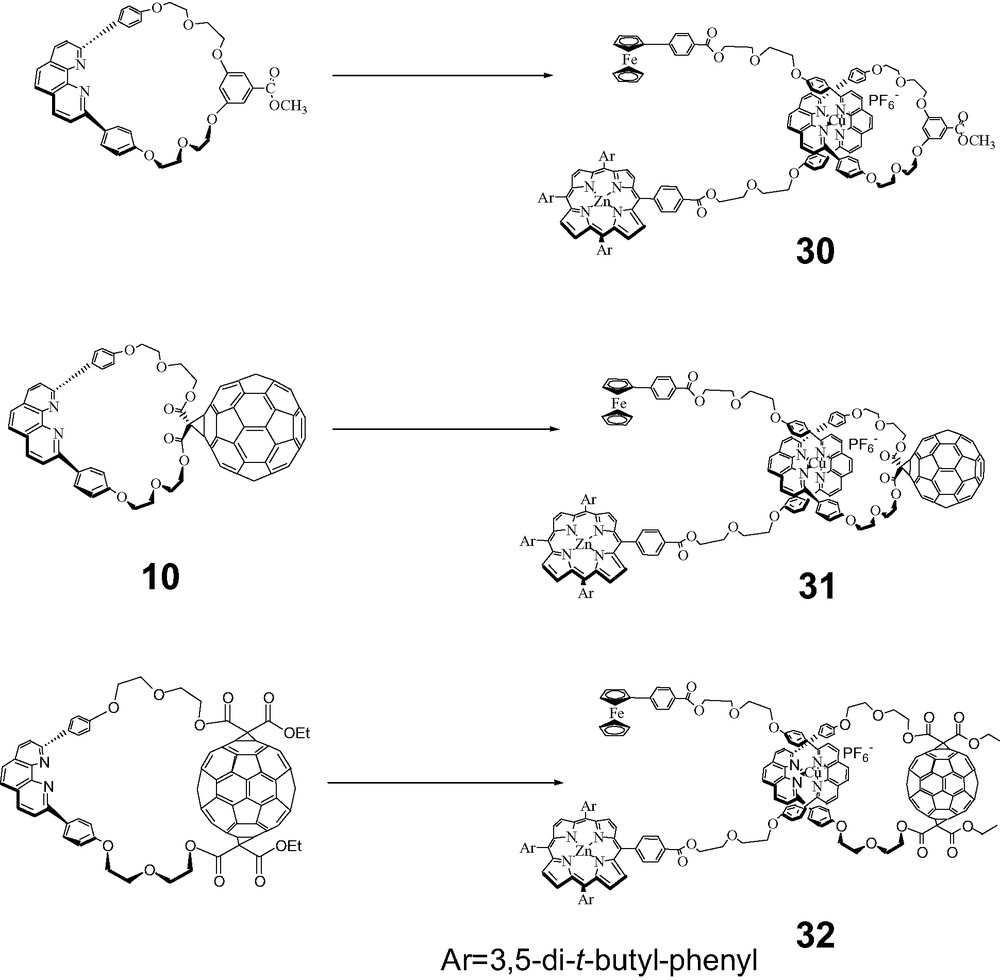
A convergent method to build rotaxanes in a single step by transition metal-assisted self-assembly is clearly much more efficient than the stepwise methods described above, and hence highly desirable. In such systems, the rotaxanes are held together only by the central transition metal complexes, and would fall apart on removal of the metal. We were indeed successful in constructing two types of rotaxanes incorporating ferrocene (Fc) moieties by self-assembly, bis-ferrocenefullerorotaxanes and zinc porphyrin/ferrocene fullerorotaxanes. In addition, Fc has a lower ionization potential than ZnP, and is consequently a better electron donor in D–A assemblies. For the first systems, the symmetrical bis-Fc phen thread 25 was prepared by the route shown in Scheme 12, by activation of Fc carboxylic acid with DIC/DMAP, which resulted in an orange solution in CH2Cl2, followed by addition of phen thread 8. The usual complexation protocol employing the same phen-containing macrocycles used earlier in the synthesis of rotaxanes 5, 6 and 7 gave the bis-Fc rotaxanes 26, 27 and 28 (see Scheme 13) [50]. For illustration, TLC analysis showed a highly fluorescent spot for macrocycle 10 and a yellow spot for 25. Complexation of 10 with Cu(MeCN)4PF6 in 3:1 CH2Cl2/MeCN followed by addition of 25 led to disappearance of both of these spots, and appearance of a new dark red spot whose MALDI-TOF mass spectrum corresponded to that of the desired rotaxane 27, indicating spontaneous self-assembly had taken place. The self-assembly process in all three cases could be readily monitored by TLC. Rotaxanes 26, 27 and 28 were isolated and purified chromatographically, and were characterized by 1H-NMR and MALDI-TOF spectral analysis.
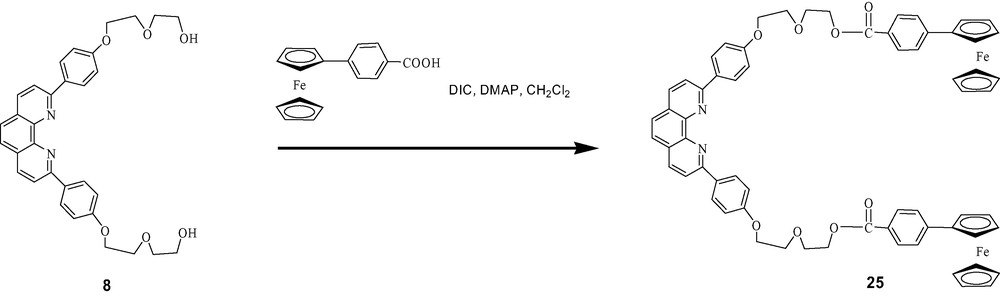
Synthesis of ferrocene-stoppered linear thread 25.
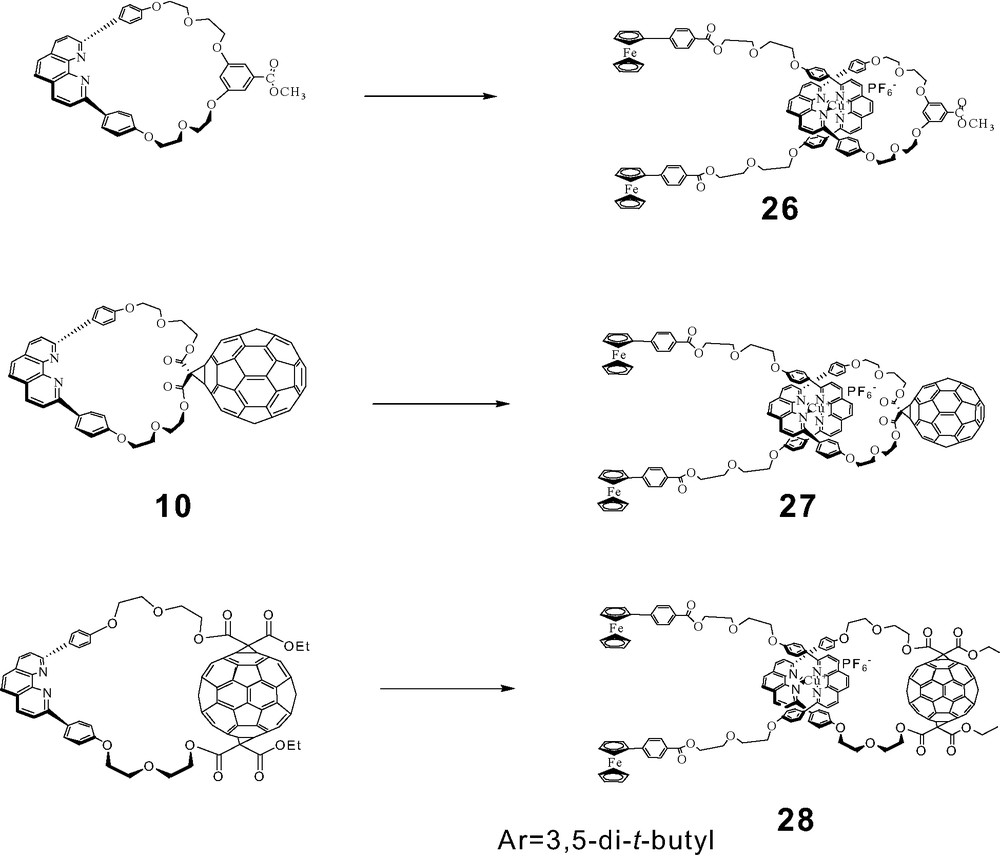
Synthesis of bis-ferrocenyl fullerorotaxanes.
A similar route was used to prepare a series of unsymmetrical fullerorotaxanes with ferrocene and porphyrin stoppers using the Fc–ZnP thread 29 shown below.
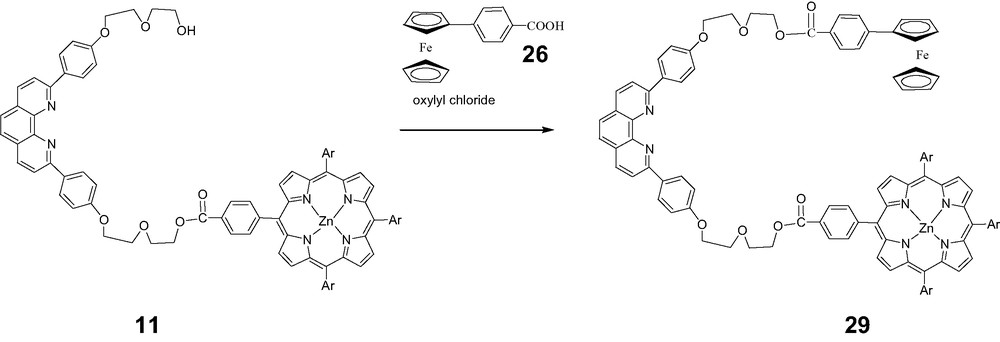
Reaction of 29 with the same set of macrocycles shown above led in one step to unsymmetrical rotaxanes 30, 31, and 32, by passage of the Fc end of the thread through the macrocycles. All products were again fully characterized by 1H-NMR and MALDI-TOF spectra [50].
9 ZnP–C60 catenane
Catenane 33 shown below was also prepared in very small quantities from the macrocyclic precursor to rotaxane 22 and a symmetrical phen thread with two terminal benzaldehyde groups, by a bis Prato 1,3-dipolar cycloaddition reaction with C60.
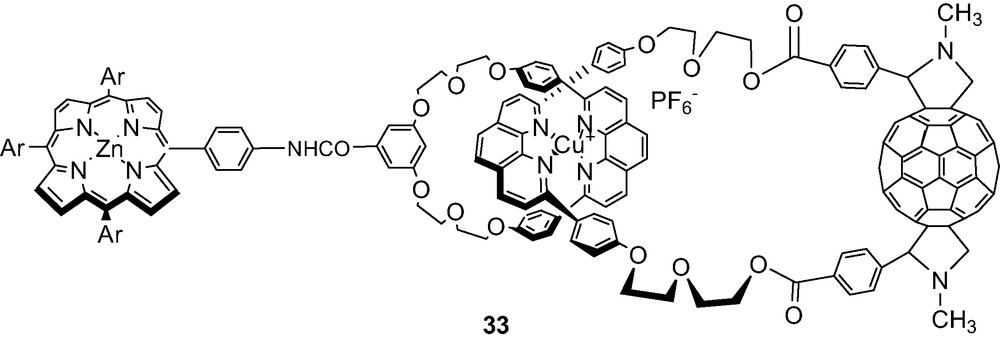
We suspect this rotaxane is a mixture of isomeric bis-adducts. This question can best be resolved by 3He-NMR analysis of the corresponding product from addition to C60 containing 3He.
10 Photophysical studies
Studies of the sequence of events following photoexcitation of the rotaxanes and catenanes described above were carried out on the femtosecond, picosecond, and nanosecond time scales in collaboration with Professor Dirk Guldi at the University of Erlangen, Germany, and Professor Helge Lemmetyinen at Tampere University of Technology in Finland. Electrochemical measurements were done by Professor Luis Echegoyen at Clemson University. These data will now be summarized. Full details will be published separately.
For the porphyrin-stoppered rotaxanes and catenanes (4,5,6,7,14,15), upon excitation of the ZnP moiety at 532 or 555 nm, steady state fluorescence and fluorescence lifetime studies indicate modest quenching of the ZnP singlet excited state. The fluorescence lifetime is reduced to ~ 1.0 ns compared with 3.2 ns for the reference zinc tetraphenylporphyrin (ZnTPP) and the fluorescence quantum yield is reduced to ~ 0.01 compared with 0.04 for the reference. This relatively slow and inefficient quenching is not typical of covalently linked porphyrin–fullerene hybrids, where fluorescence lifetimes are reduced to 10 ps or less [51]. The fact that the quenching is the same in the rotaxanes and catenanes lacking the fullerene moiety as well as those lacking C60 is a clear indication that the quenching does not involve interaction of 1ZnP* and C60. Photoinduced electron transfer (ET) to Cu(I)phen2+ is endergonic by 0.26 eV, leaving exergonic energy transfer (EnT) to the metal-to-ligand charge transfer (MLCT) state of the Cu complex as the most likely pathway. This is supported by transient absorption studies, since the charge-separated state, i.e. ZnP+●–Cu(I)phen2+–C60–●, is not detected on time scales below 1.5 ns in these systems. Excitation of the MLCT state of the Cu complex at 460 nm leads to strong quenching of the MLCT luminescence in systems lacking the C60 moiety (4 and the catenanes derived from 4) as well as those containing C60 (5,6,7 and the catenanes derived from them). However, the pathways are different. In the model system 4, exergonic EnT occurs to give porphyrin triplet states, clearly visible in transient absorption studies, while in the fullerene-containing systems ET occurs to give the charge separated state ZnP+●–Cu(I)phen2+–C60–●. The transient spectra in the latter systems show bands in the 600–800 nm range characteristic of ZnP radical cations ZnP+, and absorption at 1020–1040 nm characteristic of C60 radical anions, C60–. Decay of these transients in CH2Cl2 obeys clean unimolecular kinetics, giving lifetimes of the charge-separated radical pair (CSRP) states of 0.49, 1.17 and 0.51 μs for 5, 6 and 7, respectively. The relatively long lifetimes of the CSRP states of these rotaxanes indicates that, as with other porphyrin–fullerene hybrids, back electron transfer (BET) is occurring in the Marcus inverted region. This is supported by the stabilization of the CSRP state of bis-adduct 6 compared with monoadduct 5, since BET is more exergonic for 6 than for 5, according to electrochemical data. Since the porphyrin and fullerene moieties are not in close proximity in these systems, BET must occur through bonds by a superexchange mechanism via the Cu(I) complex. Temperature dependent studies, as well as studies in a variety of other solvents, are needed to confirm this hypothesis.
Thus, it is clear that the following sequence of events occurs following photoexcitation in these porphyrin-stoppered fullerenorotaxanes: (1) EnT on the ns time scale from 1ZnP* to the MLCT state of the Cu(I)phen2+ complex; (2) ET from the 1MLCT* to C60 to give ZnP–Cu(II)phen2++–C60–●; (3) ET from ZnP to the Cu2+ complex to give the final long-distance CSRP state ZnP+●–Cu(I)phen2+–C60–●; (4) BET to regenerate the ground state. These processes are shown schematically in Scheme 14.
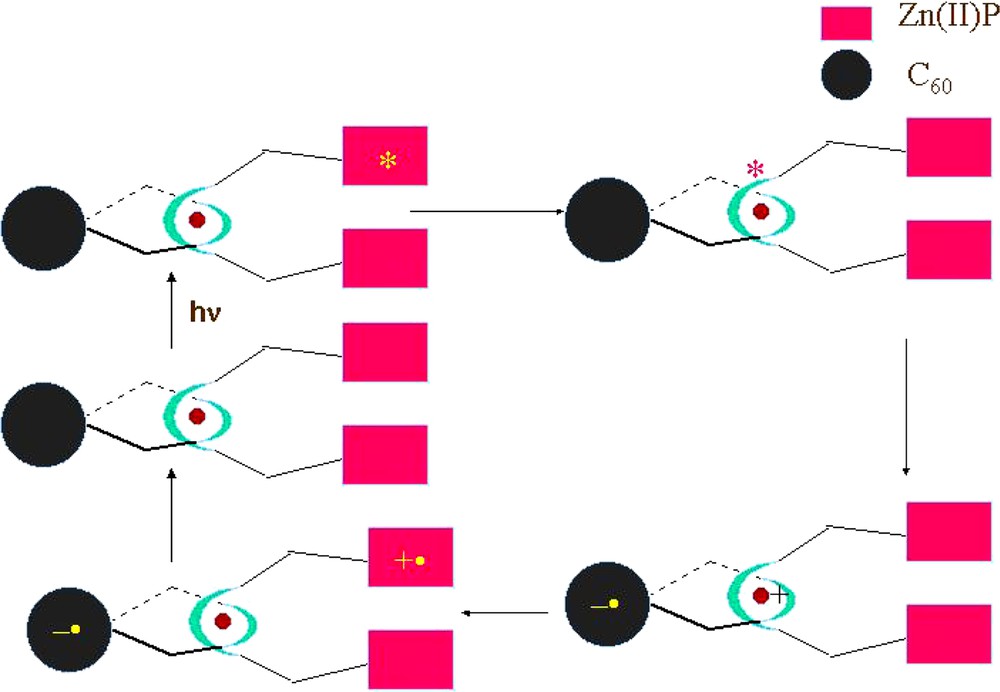
Synthesis of Fc/ZnP-stoppered fullerorotaxanes.
It is very revealing that the dynamics following photoexcitation of catenanes 14 and 15 are nearly identical to that of the precursor rotaxane 5 (the photophysics of the other catenanes has not yet been studied). These include the lifetimes of the 1ZnP* states, the transient absorption spectra and, most importantly, the lifetimes of the CSRP states. Thus, decay of the ZnP●+ / C60●– radical ion pair transient absorption for rotaxane 2, and catenanes 14 and 15 in CH2Cl2 all show clean first-order kinetic behavior, corresponding to CSRP lifetimes of 500 ± 50 ns for all three materials. This can be rationalized in terms of a pre-organized structure for rotaxanes 5, 6, and 7, which provides a molecular scaffold for binding of DABCO and 4,4′-bipyridyl without inducing significant topological changes. This result is quite different from a previously studied system where bridging between two ZnP groups using DABCO results in a drastic change in the conformation and photophysical properties of the system [52].
An energy level diagram for our systems is shown in Fig. 5.
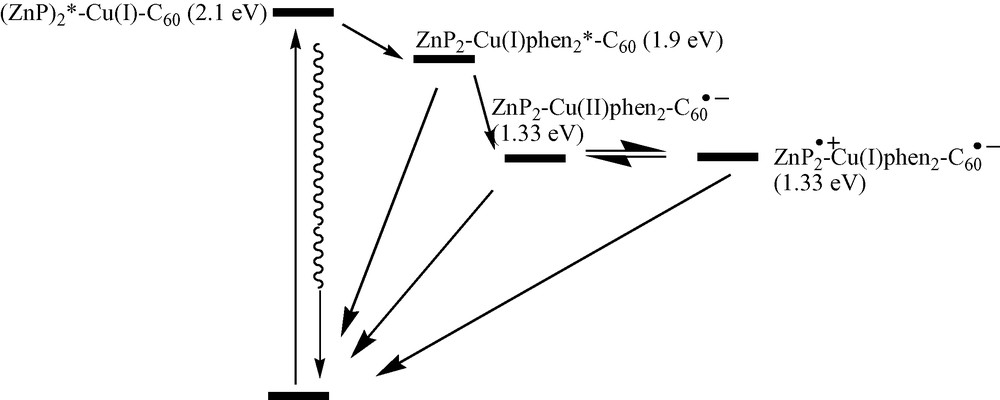
Energy diagram for porphyrin-stoppered fullerorotaxanes and fullerocatenanes.
Qualitatively similar photophysical results were obtained for the fullerene-stoppered rotaxanes 20, 21 and 22. For the model rotaxane 20, transitions in the ns time range attributable to formation of the charge-separated state Cu(phen)22+-C60●– following photoexcitation of the fullerene moiety were observed, with transient absorption at 1000 nm. The lifetime of this state in CH2Cl2 was 320 ns. The ZnP fluorescence lifetimes of 21 and 22, 0.22 and 0.44 ns, respectively, were identical with those of the synthetic precursors before attachment of the fullerene moieties; all were shorter than that of the ZnP reference (3.2 ns). This clearly shows that, as in the rotaxanes and catenanes discussed earlier, 1ZnP* is interacting directly with the Cu(phen)2+ moiety and is not ‘talking to’ the fullerene. The shorter singlet state lifetime of 21 and its precursor vis a vis 22 and its precursor is attributed to two factors: (1) the reduced separation of the interacting groups in 21 compared to 22, and (2) differences in topology. In 21, the planes of the ZnP and ‘western’ phen groups are essentially coplanar, while they are nearly perpendicular in 22 because of the extra phenyl group in the linker. The relative importance of these two factors remains to be evaluated.
The MLCT luminescence in 21 and 22 is barely detectable due to rapid intramolecular ET to the fullerene. Thus the MLCT lifetime of 21 and 22 is 0.58 and 0.59 ns, respectively, compared to 58 ns for a reference pseudorotaxane lacking fullerene and porphyrin moieties. Both 21 and 22 ultimately give CSRP states ZnP+●-Cu(I)phen2+-(C60)2–● with transient absorption features as described above. The decay of both the ZnP+● and C60–● absorptions obey clean first order kinetics, corresponding to CSRP lifetimes for 21 and 22 of 0.73 and 29 μs in CH2Cl2, and 0.89 and 32 μs in THF, respectively. These values for 22 are among the longest ever reported for the CS state of a simple porphyrin–fullerene dyads as contrasted with multicomponent fullerene–porphyrin arrays where central free base porphyrins act as ET relays between terminal ZnP and C60 moieties. The overall sequential photophysical pathway in these systems is totally analogous to that shown in Scheme 14. The long CSRP lifetime of 22 could be due to a variety of factors, including distance, the chemical nature of the linker, and molecular topology, all of which need to be individually evaluated. It is clear at this point, however, that relatively small structural modifications can have a large effect on the photodynamics of these systems. We believe that even longer CSRP lifetimes can be achieved using these molecular architectures.
For study of ET processes in the Fc-stoppered rotaxanes 26, 27 and 28, the MLCT excited states of the Cu(I)phen2+ moiety was excited at 460 nm, where the fullerene also absorbs to some extent. The steady-state emission spectra shown in Fig. 6 demonstrate quenching of the MLCT emission in the fullerorotaxanes 27 and 28 relative to the model compound 26. Interestingly, the fluorescence lifetime of 26 is shorter than that of the precursor pseudorotaxane lacking the Fc groups, due either to a heavy atom effect or EnT from the MLCT state to the Fc. Because the Fc group is optically transparent, we cannot distinguish between these alternatives. The MLCT luminescence lifetimes of 27 and 28, 0.25 and 0.30 ns, respectively, are shorter than those of the rotaxanes discussed earlier, for reasons which are not clear at this time. Quenching is attributed to exergonic photoinduced ET from the lowest MLCT state (1.70 eV above the ground state) to generate the CS state Cu(II)phen2–C60●– at 1.33 eV, from electrochemical data. Formation and decay of the C60 radical anion could be followed from the absorption at 1000 nm. Fullerenorotaxane 34, which lacks the Fc groups, has a CS lifetime of 140 ns in CH2Cl2, while the CS lifetimes for 27 and 28 are 1170 and 1010 ns, respectively. In the latter systems, charge shift from Fc to Cu(II), which is energetically downhill, leads to long-distance CS states Fc+●–Cu(I)phen2+–C60●–. In these systems, we postulate that BET is probably in the normal reason of the Marcus inverted region, since the thermodynamic driving force –ΔG0BET for 27 is smaller than that for 28, from electrochemical data, while the CS lifetime is 27 is slightly longer than that of 28. Since the difference is not large, this conclusion must be regarded at this time as tenuous. The photophysical pathways which ensue on photoexcitation of 27 are summarized in Fig. 7.
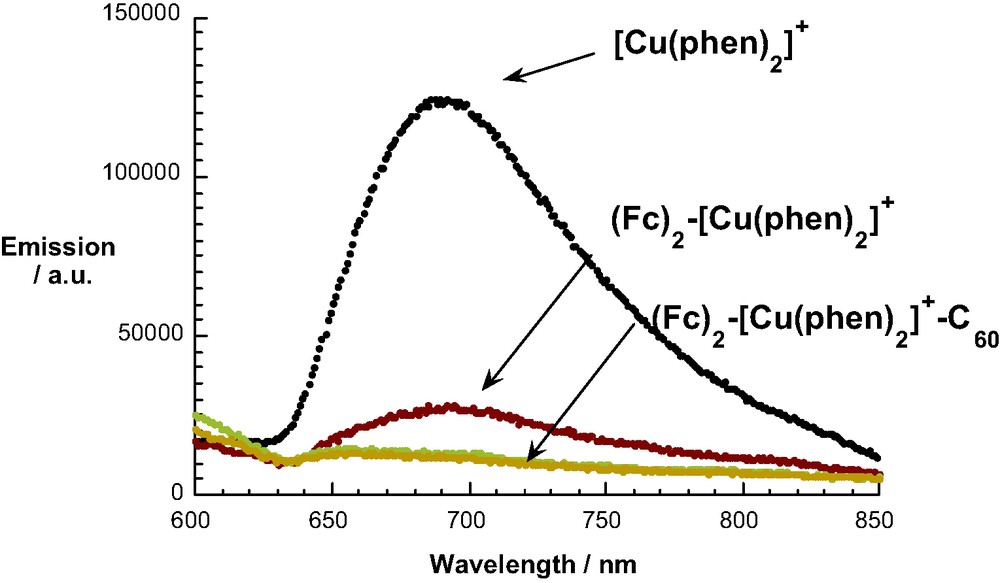
MLCT emission in Fc-stoppered rotaxanes.
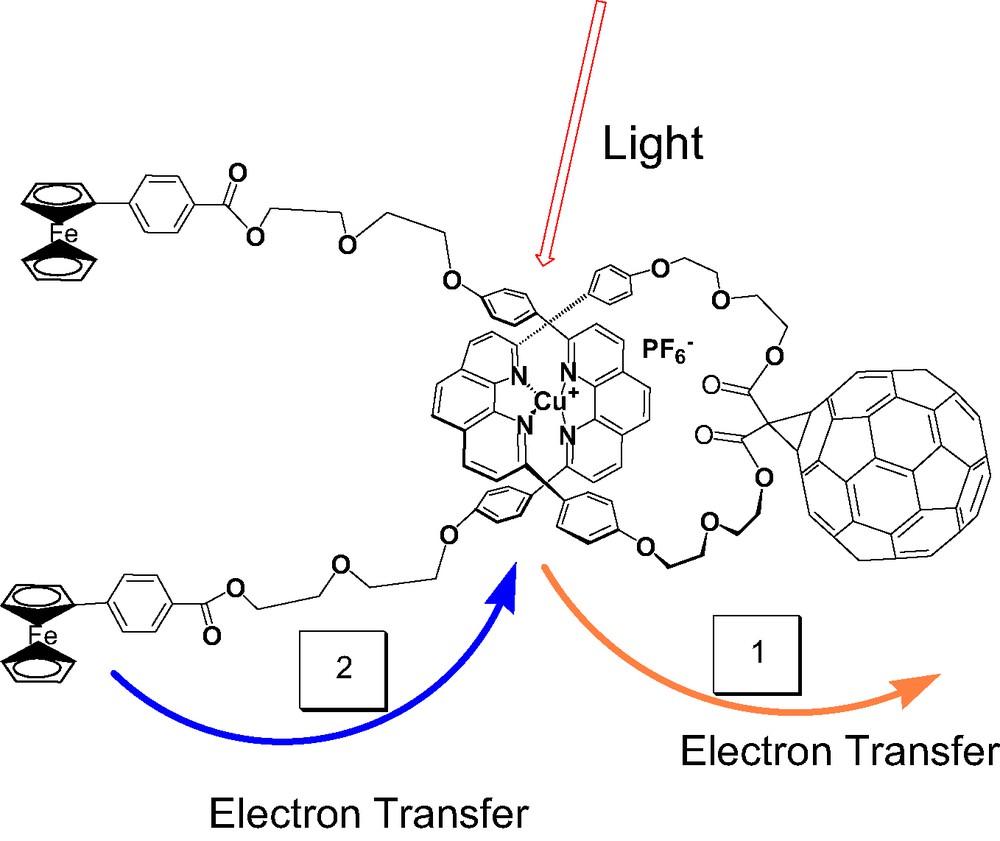
Photophysical pathways for Fc-stoppered fullerorotaxane 28.
With the ZnP/Fc-stoppered rotaxanes 30, 31 and 32, we again have a chromophore, namely ZnP, that absorbs strongly in the visible, and whose behavior as an electron donor can be followed spectroscopically. In these systems, there is a competition between Fc and ZnP as electron donors toward the oxidized copper complex generated by the routes discussed earlier. In systems studied by Imahori et al., involving linear Fc-ZnP–H2P–C60 tetrads and related materials, Fc acts as the ultimate electron donor. For rotaxanes 31 and 32, the transient absorption spectra are similar to those obtained earlier, namely showing absorption characteristic of ZnP+● and C60●–. The CSRP lifetimes obtained by monitoring transient decay are 0.64 and 1.3 ms, respectively, which are not significantly larger than those of the bis-ZnP-stoppered fullerorotaxanes 5 and 6, respectively, indicating little, if any, effect of the Fc moiety. Whether or not Fc+ is formed, either by through bond ET to the oxidized copper or through space ET to oxidized ZnP, both of which are energetically favorable, is impossible to tell with certainty from our spectroscopic data. If such processes did occur, enhancement in CSRP lifetime would be expected, and this is not observed. Examination of these systems using time-resolved electron paramagnetic resonance (TREPR), in which it should be possible to detect Fc+●, should provide an answer to this question. This technique has recently been used to elucidate photophysical pathways in other porphyrin–C60 hybrid materials [53].
While extensive photophysical studies of catenane 33 have not been carried out as yet, we have determined that its CSRP lifetime in THF is 14 μs, indicating BET is quite slow. It will be very interesting to see how much this can be extended by attaching an Fc moiety to the porphyrin, further stabilizing the cationic component while simultaneously increasing the distance between the charged moieties.
As mentioned earlier, the topology and hence the photophysics of these systems, should change drastically on removal of Cu(I) from these rotaxanes and catenanes. Experiments along these lines are currently in progress. The method used by Sauvage, namely treatment with KCN, is inapplicable in the systems already possessing C60 because of nucleophilic attack on the fullerene. New demetallation protocols need to be developed. We do not expect this to be an insurmountable problem.
Acknowledgments
Our research in this field has been supported by grants from the U.S. National Science Foundation and the Petroleum Research Fund, administered by the American Chemical Society. Support is also gratefully acknowledged from a fund established to support research in the Schuster laboratory from former members and friends of the group. K.L. also acknowledges a MacCracken fellowship from the Graduate School of Arts and Science at NYU. The NYU major instrument facility was established in part by grants from the National Science Foundation under their major instrument program. D.M.G. gratefully acknowledges support from the Office of Basic Energy Sciences of the U.S. Department of Energy, SFB 583, and Fonds der Chemischen Industrie.